







Cheng, Y., Su, Y., Yu, Z., Liang, Y., Wong, K.-C., & Li, X. (2023). Unsupervised Deep Embedded Fusion Representation of Single-Cell Transcriptomics. Proceedings of the AAAI Conference on Artificial Intelligence, 37(4), 5036-5044. https://doi.org/10.1609/aaai.v37i4.25631
写在前面
这篇论文和上一篇论文阅读:ZINB-Based Graph Embedding Autoencoder for Single-Cell RNA-Seq Interpretations-CSDN博客
分享的论文有共同的通讯作者,具有一定的连贯性,因此放在一起学习。
摘要
细胞聚类是分析单细胞RNA测序(scRNA-seq)数据的关键步骤,它允许在单细胞水平上进行转录谱分析后对细胞异质性进行表征。最近,单细胞深度嵌入表示模型因其可以同时学习特征表示和聚类而受到青睐。然而,这些模型仍然面临着许多重大挑战,包括海量数据、普遍的dropout事件以及转录谱中复杂的噪声模式。
本文提出了一种名为单细胞深度嵌入融合表示(scDEFR)模型,它产生了一个深度嵌入融合表示,以学习包含基因水平转录组和细胞拓扑信息的融合异质潜在嵌入。首先逐层融合它们,以获得细胞间关系和转录组信息的压缩表示。然后,使用基于零膨胀负二项式模型(ZINB)的解码器来捕获数据的全局概率结构,以重构最终的基因表达信息。最后,通过同时集成聚类损失、交叉熵损失、ZINB损失和细胞图重构损失,scDEFR可以在联合互相监督策略下优化聚类性能,并从融合信息中学习潜在表示。论文对来自不同测序平台的 15 个单细胞RNA-seq 数据集进行了全面的实验,并证明了scDEFR相对于各种最先进方法的优越性。
写作动机
单细胞RNA测序(scRNA-seq)技术允许分析大量个体细胞的整个转录组,这对于表征细胞群体内的随机异质性并建立不同的发育轨迹至关重要,例如,免疫细胞。 (Papalexi和Satija 2018)。由于基因组覆盖的高度异质性和技术限制,scRNA-seq数据非常稀疏,其中95%的测量值为零(Grün,Kester和Van Oudenaarden 2014)。这些问题阻碍了依赖相似性度量的传统聚类方法的充分适用。基于自编码器AE的scRNA-seq聚类方法虽然解决了上述问题,但是缺少了对相邻细胞信息的分析利用,而基于图自编码器GAE的scRNA-seq聚类又会缺少关键的基因表达数据。因此,将两种方法进行融合,使得可以从基因表达矩阵和细胞图中同时学习潜在表示。
创新点
- 提出了一种称为scDEFR(a Single-Cell Deep Embedding Fusion Representation model)的单细胞深度嵌入融合表示模型,将细胞拓扑编码器与基于转录组谱的图编码器结合起来,以捕获scRNA-seq数据的融合压缩表示。
- scDEFR采用TAGCN从数据中提取拓扑信息,然后在编码过程中跨越编码器的每一层进行融合增强。此外,scDEFR利用基于ZINB的多模态融合解码器捕获数据的全局概率结构,对高度稀疏和过分分散的scRNA-seq数据进行建模。
- 据本文所知,这是首个提出融合异质结构信息来解决单细胞转录组学分析的架构。
- 将本文的方法与15个真实的scRNA-seq数据集上的竞争性最新方法进行了比较。结果表明,scDEFR优于所有其他基线方法。
模型
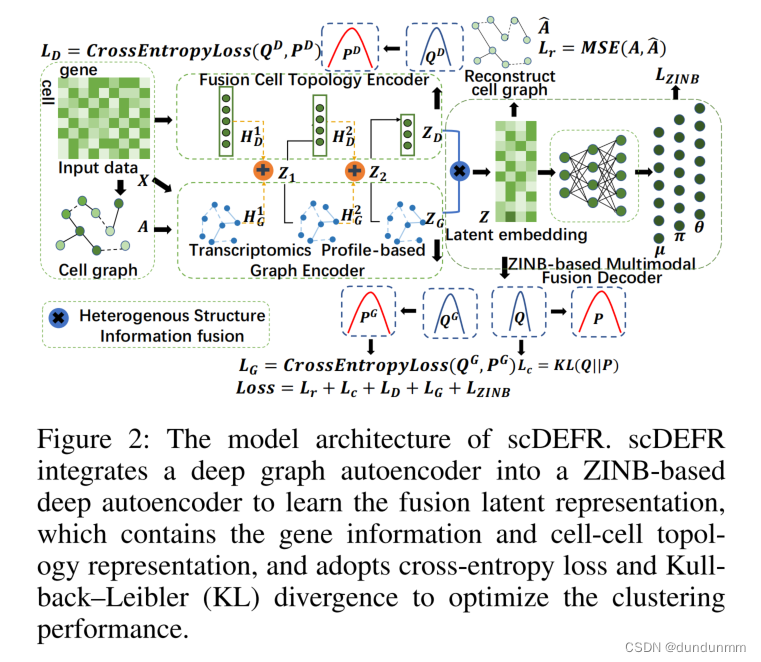
1、融合细胞拓扑编码器
大多数先前的深度嵌入式聚类方法通常专注于学习基因表达矩阵,而忽略了细胞之间的拓扑结构。因此,在本文的研究中,将细胞间的拓扑结构嵌入到一个侧重于基因级别信息分析的编码器中。为了将细胞拓扑信息融合到基因级别的有效特征表示中,提出了一个具有层间嵌入操作的融合细胞拓扑编码器。融合细胞拓扑编码器由三个全连接层组成。与先前的研究相比,本文将融合后的压缩表示视为全连接层的输入,并将其与每一层的细胞间拓扑结构信息相结合。融合细胞拓扑编码器第l层的输入可以表述如下:
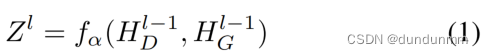
其中,是两种不同隐藏信息的线性组合,
是预定义的超参数;HD是融合细胞拓扑编码器中第 l-1 层的隐藏信息,HG是基于转录组文件的图编码器中第 l-1 层的隐藏信息。在此之后,融合细胞拓扑编码器中的潜在表示可以如下获得:
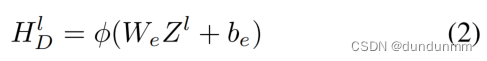
其中,是ReLU(),即全连接层的激活函数;
和
分别是融合细胞拓扑编码器的可学习权重矩阵和偏置。
2、基于转录组文件的图编码器
现有的单细胞图嵌入自编码器缺乏一种稳健而协同的方法,用于嵌入细胞拓扑和基因级别的转录组信息,以进行特征表示学习。为了捕捉基因表达数据的特征表示以及细胞之间的关系,本文开发了一个基于转录组文件的图编码器,将基因级别的转录组信息整合到图编码器中。模型中的基于转录组文件的图编码器由三个拓扑自适应图卷积层(TAGCN)组成(Du等人,2017)(详见论文阅读:ZINB-Based Graph Embedding Autoencoder for Single-Cell RNA-Seq Interpretations-CSDN博客)。拓扑自适应图卷积层使用M个图卷积核来提取不同尺寸的局部结构特征,可以完全提取图信息。多项式卷积核的内部架构定义如下:
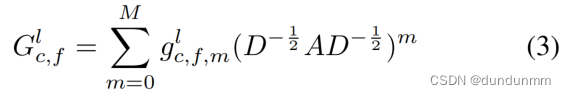
为了将来自融合细胞拓扑编码器的基因水平转录组信息 嵌入到基于转录组文件的图编码器中,使用压缩表示
作为TAGCN在第l个隐藏层的输入特征图,该特征图由以下公式计算得出:
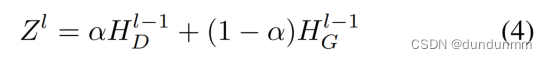
在此基础上,集成表示可以用于生成一个新的表示,用于学习基因表达数据的特征表示和细胞之间的关系,为融合的基因表达表示提供了近似的二阶图正则化(Bo等人,2020)。特别是,我们假设在进行特征映射之前,每个节点具有 个特征,第c个特征为
,其中
。然后,TAGCN的卷积操作过程定义如下:
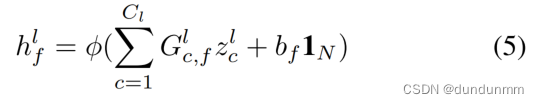
为了简化,可以给出基于转录组文件的图编码器输出的表述:
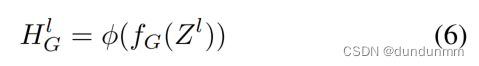
5、异构结构信息融合
受跨模态融合机制的启发(Tu等人,2021),本文通过整合融合细胞拓扑编码器和基于转录组文件的图编码器的两个潜在嵌入(ZD和ZG),生成融合的异构潜在表示Z。事实上,本文的模型考虑了局部和全局细胞之间的相关性。在经过基于转录组文件的图编码器处理细胞图之后,编码器的潜在嵌入将包含各种结构信息的顺序。然后,将它们与转录组基因级别表示融合,从而减少具有多跳关系的细胞之间的相关性。
6、基于ZINB多模态融合解码器
在将来自融合细胞拓扑编码器和基于转录组学档案的图编码器的压缩表示合并后,获得了作为解码器输入的融合异构潜在嵌入Z。与其他融合模型不同,本文采用了基于零膨胀负二项式(ZINB)模型的单个解码器,同时连接两个编码器模型,旨在重构单细胞转录组档案和细胞图。 ZINB分布用于捕获数据的整体概率结构,特别适用于建模高度稀疏和过度分散的基因表达数据。基于此,提出了一种基于ZINB的多模态融合解码器,以捕获单细胞RNA测序数据的特征。
最初,重构\(\overline{X}\)的解码器可以表示为:
\[
\overline{X} = f_{\text{dec}}(W'Z + b')
\]
这里,\(W'\)和\(b'\)分别表示解码器的权重矩阵和偏置向量;\(f_{\text{dec}}\)表示一个三层全连接神经网络,\(Z\)是通过异构结构信息融合机制获得的融合潜在嵌入。
重构的邻接矩阵\(A_r\)定义为潜在嵌入的内积:
\[
A_r = \sigma(Z^T Z)
\]
将\(A\)的重构损失\(L_r\)定义为:
\[
L_r = \| A - A_r \|_2^2
\]
随后,为了捕获数据的全局概率结构,将基于ZINB的解码器集成到我们的解码器模型中。这将三个独立的全连接层与最后一层连接起来,以估计ZINB的参数:丢失率\(\pi\),离散度\(\theta\)和均值\(\mu\)。网络输出的参数矩阵定义如下:
\[
\Pi = \text{sigmoid}(W_{\pi} \overline{X})
\]
\[
M = \exp(W_{\mu} \overline{X})
\]
\[
\Theta = \exp(W_{\theta} \overline{X})
\]
这里,\(W\)表示损失函数的学习权重。基于ZINB的解码器重构了单细胞RNA测序数据,如下所示:
\[
\text{NB}(X|\mu, \theta) = \frac{\Gamma(X + \theta)}{X! \Gamma(\theta)} \left( \frac{\theta}{\theta + \mu} \right)^{\theta} \left( \frac{\mu}{\theta + \mu} \right)^X
\]
\[
\text{ZINB}(X|\pi, \mu, \theta) = \pi \delta_o(X) + (1 - \pi) \text{NB}(X)
\]
然后,原始数据\(X\)的重构损失函数定义为ZINB分布的负对数似然:
\[
L_{\text{ZINB}} = -\log(\text{ZINB}(X|\pi, \mu, \theta))
\]
7、联合互相监督策略
由于深度嵌入聚类方法是无监督的,本文设计了一种互相监督的策略,将融合细胞拓扑编码器、基于转录组学文件的图编码器和聚类模块统一到一个统一的优化框架中,以有效地训练这两个模块进行聚类。在模型中,它由三个不同的分布块组成:融合模型scDEFR的聚类分布Q、目标分布P和融合异构潜在嵌入Z;融合细胞拓扑编码器的聚类分布QD、目标分布PD和潜在嵌入ZD;以及基于转录组学档案的图编码器,具有聚类分布QG、目标分布PG和潜在嵌入ZG。这三个相同块中的分布是相互监督的,并且统一在一个框架中进行学习和训练。考虑到融合模型scDEFR,定义了软标签qiu如下:
\[
q_{iu} = \left(1 + \frac{\|z_i - \mu_u\|^2}{1 + \frac{\|z_i - \mu_r\|^2}{P_r}}\right)^{-1}
\]
该标签表示潜在嵌入zi与聚类中心μu之间的相似性,μu是在基于ZINB的多模态融合解码器预训练后通过谱聚类或K-Means聚类生成的。此外,基于qiu,定义了辅助目标分布piu:
\[
p_{iu} = \frac{q_{iu}^2/\sum_i q_{iu}}{\sum_r q_{ir}^2/\sum_i q_{ir}}
\]
最后,通过最小化聚类目标(Xie, Girshick, and Farhadi 2016)来采用Kullback-Leibler(KL)散度,定义如下:
\[
L_C = \text{KL}(P||Q) = \sum_i \sum_u p_{iu} \log \frac{p_{iu}}{q_{iu}}
\]
可以看出,分布P监督分布Q的学习,目标分布P是由分布Q计算得到的。这种互相监督策略有助于为融合模型学习更好的数据表示,从而实现更高质量的聚类。
此外,为了使不同模型生成的潜在表示尽可能接近原始数据的聚类中心,并避免整个模型的崩溃,使用二元交叉熵作为另一个目标函数,使用分布PD和PG来分别监督分布QD和QG。注意,它们是通过使用计算p和q的等式,并将Z替换为ZD和ZG来计算的。因此,目标分布可以帮助编码器学习更好的潜在表示以获得更好的聚类结果。它由一个融合细胞拓扑编码器和一个基于转录组学文件的图编码器生成,如下所述:
\[
L_D = -P_D \log(Q_D) - (1 - P_D) \log(1 - Q_D)
\]
\[
L_G = -P_G \log(Q_G) - (1 - P_G) \log(1 - Q_G)
\]
因此,在训练过程中,融合细胞拓扑编码器和基于转录组的图编码器以及它们的融合表示的潜在表示将与强大的目标分布同时对齐。scDEFR方法有五个优化目标:
\[
L = \gamma_1 L_r + \gamma_2 L_{ZINB} + \gamma_3 L_C + \gamma_4 (L_D + L_G)
\]
其中,\(L_r\)是重构损失;\(L_{ZINB}\)是ZINB损失;\(\gamma_1, \gamma_2, \gamma_3\)和\(\gamma_4\)是权重系数,用于控制总损失函数的平衡。
实验
1、实验数据
为了证明scDEFR的有效性,将本文的方法应用于来自(Yu et al. 2022)的十五个真实单细胞RNA测序数据集。这十五个真实数据集来自七种不同的代表性测序平台,涵盖了多个物种。详细信息如表1所述。首先过滤掉在超过1%的细胞中表达为非零的基因和不表达的基因。其次,使用scanpy包对数据进行了归一化处理。接下来,基于归一化值的排名选择了前d个高变量基因。最后,采用KNN算法构建了细胞图,其中图中的每个节点代表一个细胞。对于每个细胞,识别其前K个相似邻居,并通过边将它们连接起来。

2、实验设置
使用最近邻参数为K = 10的KNN算法构建了细胞图。此外,使用了结合的融合细胞拓扑编码器和基于转录组文件的图编码器来构建网络,并将线性融合参数α设置为0.1;每一层配置了1024、128和24个节点;全连接解码器的层采用了对称编码器形式。特别地,算法包括预训练和训练两个阶段,都设置为250个epochs。Adam算法被用作优化器,预训练的学习率为5e-5,正式训练的学习率为1e-7。目标函数的权重系数{γ1, γ2, γ3, γ4}分别设置为{0.3, 0.1, 0.5, 0.1}。基线方法的参数设置与原始出版物完全相同。最后,实验在一台搭载NVIDIA Quadro RTX 6000 GPU和24GB内存的Ubuntu服务器上进行。
3、实验结果
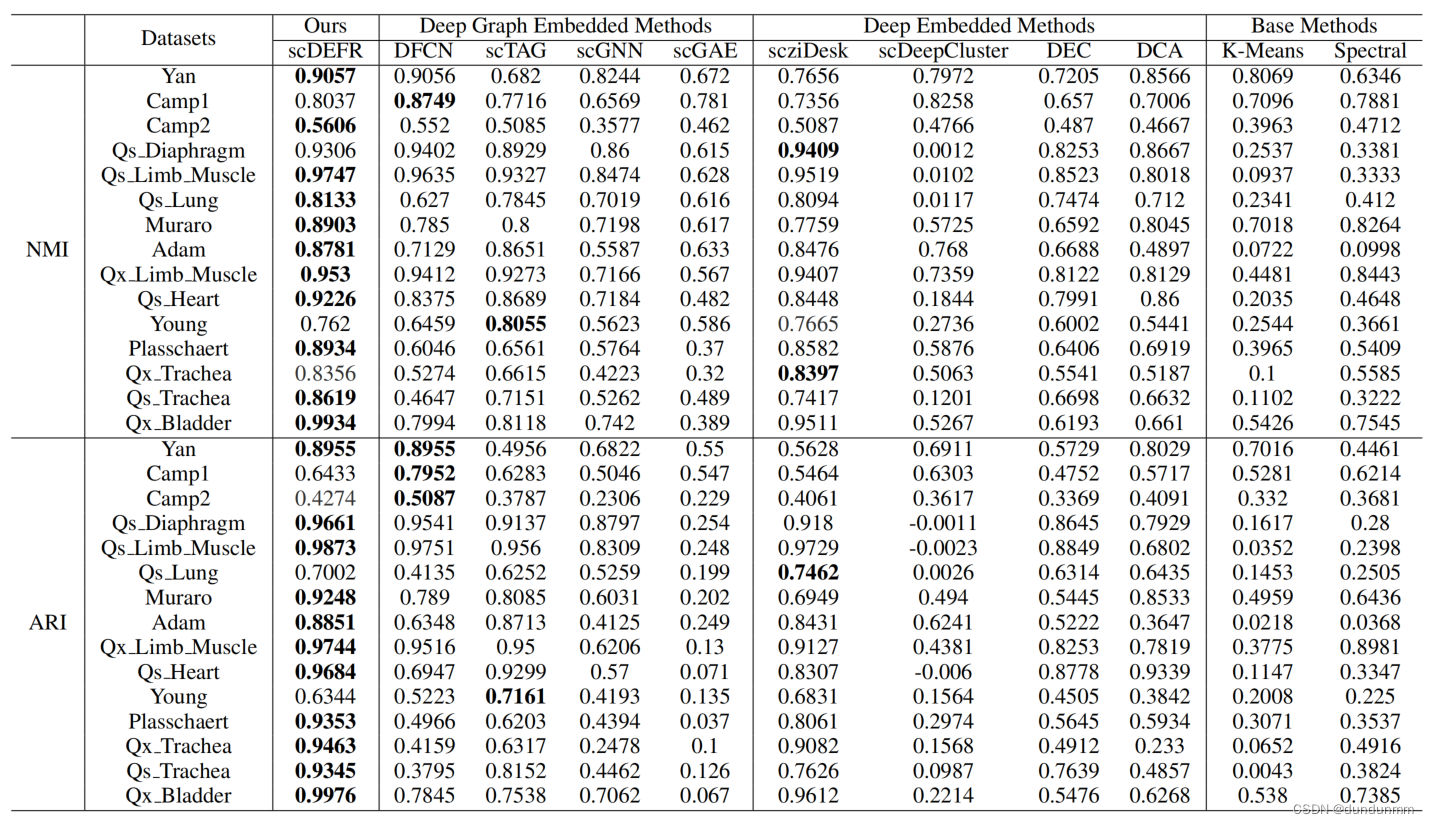
论文的评测指标为聚类的指标,虽然论文题目没有包含聚类关键字,但是主要还是利用聚类来识别细胞类别。
细胞的异质结构信息融合这个创新点的名字提的不错,赞一下















 669
669

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









