








摘要:构建具有微配位环境的单原子催化剂是实现令人满意的催化性能的一种有前途的策略。本文通过简单的煅烧温度控制策略,成功制备了具有精确控制配位环境的铂单原子负载在 CeO2 催化剂。实验和理论计算相结合表明,在 550 °C(Pt/CeO2-550)下制备的 Pt1/CeO2上的铂单原子主要位于 CeO2的边缘位点,Pt-O 配位数约为 5;而在 800 °C(Pt/CeO2-800)下制备的 Pt1/CeO2上的铂单原子主要位于 CeO2 台面上的扭曲 Ce 取代位点,Pt-O 配位数约为 4。具有不同 Pt1-CeO2 配位环境的 Pt/CeO2-550 和 Pt/CeO2-800 由于在反应物活化和 H2O 解吸中的不同优势,在 CO 氧化和 NH3 氧化中表现出相反的活性趋势。
介绍
近年来,不同类型的单原子催化剂因其最大的原子利用效率、独特的电子状态或与纳米团簇/粒子催化剂相比更高的稳定性,已被应用于许多与能源和环境相关的反应中。随着该领域研究的不断深入,人们发现,通过调整单原子催化剂的氧化态或配位环境,可以显著提高其在特定载体上进行某些反应(如热催化和电催化)的性能。在这些单原子催化材料中,以稀土金属氧化物 CeO2 为载体的铂单原子(Pt1)催化剂是最受欢迎的催化剂体系之一,已被广泛研究用于消除环境污染物和能源转换。
最近,人们采用了各种合成方法来制造催化性能更好的铂单原子。例如,通过 750 °C 水热处理在 Pt1/CeO2 催化剂上形成稳定的羟基,可显著提高 Pt1/CeO2 上的 CO 氧化活性。Ma 等人1报道用磷酸盐(PO43-)调节 Pt1/CeO2 可以提高铂的价态,促进反应物的吸附和氢的溢出,从而提高苯乙烯氢化的催化活性。Jeong 等人2利用氢还原法调整了 CeO2 上铂单原子的氧化态,以最大限度地提高 CO、CH4 和 NO 氧化的催化活性。与此同时,许多实验和理论工作者致力于在 CeO2 载体上合成具有最佳配位环境的 Pt1 催化剂,或构建合理的 Pt1-CeO2 结构模型,这有助于更好地理解反应机理,并揭示铂单原子催化剂不同催化性能的根源。例如,Jiang 等人3成功合成了具有不对称 Pt1-O4 构型的 Pt1/CeO2 催化剂,其 CO 氧化活性远高于具有对称 Pt1-O4 构型的参考催化剂。Wang 等人4报告发现CO-Pt1-O3 是氧等离子体预处理 CeO2 上负载的 Pt1 催化剂吸附 CO 的主要构型,与传统 Pt1/CeO2 催化剂相比,这种 Pt1 物种表现出更高的 CO 氧化活性和更好的抗烧结性。迄今为止,还没有全面的研究报告关注如何精确调节 CeO2载体上铂单原子的局部配位环境,以调节其在不同催化反应中的活性,同时揭示其内在的结构-活性关系。在此,我们提出了一种简单的策略,即通过控制简便的初湿浸渍(IWI)法制备的 Pt/CeO2 催化剂的煅烧温度来微调铂单原子的精确位置。在不同温度下煅烧的 Pt1/CeO2催化剂上的铂单原子可能具有不同的配位环境。通过对 Pt1/CeO2 催化剂进行系统的实验表征、催化性能测试和基于密度泛函理论(DFT)的模拟,可以清楚地发现具有微调局部配位结构的 CeO2 负载的铂单原子在不同的氧化反应(如 CO 氧化和 NH3氧化)中可以表现出截然不同的催化行为,这需要在未来单原子催化的实际应用中加以考虑。
结果
利用不同的协调环境构建 Pt1单原子
为了尽量减少纯 CeO2 载体在沉积Pt并煅烧后可能发生的结构变化,本研究中使用的 CeO2 在 800 °C 的空气中预煅烧 12 小时,以获得预稳定结构。此外,有报道称铂的沉积对稳定 CeO2 载体有积极作用。如补充图 1 和表 1 所示,纯 CeO2 载体和 CeO2负载的铂催化剂显示出几乎相同的比表面积和孔结构,表明在催化剂制备过程中 CeO2 载体的质构没有发生明显变化。
 为了确定Pt/CeO2催化剂上Pt物质的状态,首先在25℃下进行CO吸附的原位DRIFTS(图 1)。在所有四种催化剂上均在 2087–2105 cm-1处观察到分配给吸附在离子 Pt 单位点上的 CO (CO-Ptδ+@Pt1)的红外谱带。对于 Pt/CeO2-350,在ca处有一个宽带。还检测到2050 cm-1归因于吸附在来自Pt簇的金属Pt位点上的CO(CO-Pt0@Pt簇)。虽然Pt/CeO2 -550、Pt/CeO2-700和Pt/CeO2-800上的Pt物种主要以单原子形式存在,但CO-Ptδ+ @Pt1带的单调红移为煅烧温度的升高表明Pt单原子的局部配位环境或氧化态不同。根据之前的工作得出的结论,分配给线性吸附在由CeO2负载的Pt1上的CO的IR谱带中心通常在2085和2105 cm-1 之间。随着煅烧温度从550℃升高到800℃,CO吸附的IR谱带红移(从2096到2087cm-1) 9 cm-1可以被认为是一个显着的变化,表明Pt上明显不同的Pt1局域结构/CeO2 -550 和 Pt/CeO2-800。此外,在不同温度下煅烧的Pt/CeO2催化剂显示出不同的颜色(从深棕色到金棕色),表明Pt1物种也存在不同的状态(插图 1a-d中的照片)。
为了确定Pt/CeO2催化剂上Pt物质的状态,首先在25℃下进行CO吸附的原位DRIFTS(图 1)。在所有四种催化剂上均在 2087–2105 cm-1处观察到分配给吸附在离子 Pt 单位点上的 CO (CO-Ptδ+@Pt1)的红外谱带。对于 Pt/CeO2-350,在ca处有一个宽带。还检测到2050 cm-1归因于吸附在来自Pt簇的金属Pt位点上的CO(CO-Pt0@Pt簇)。虽然Pt/CeO2 -550、Pt/CeO2-700和Pt/CeO2-800上的Pt物种主要以单原子形式存在,但CO-Ptδ+ @Pt1带的单调红移为煅烧温度的升高表明Pt单原子的局部配位环境或氧化态不同。根据之前的工作得出的结论,分配给线性吸附在由CeO2负载的Pt1上的CO的IR谱带中心通常在2085和2105 cm-1 之间。随着煅烧温度从550℃升高到800℃,CO吸附的IR谱带红移(从2096到2087cm-1) 9 cm-1可以被认为是一个显着的变化,表明Pt上明显不同的Pt1局域结构/CeO2 -550 和 Pt/CeO2-800。此外,在不同温度下煅烧的Pt/CeO2催化剂显示出不同的颜色(从深棕色到金棕色),表明Pt1物种也存在不同的状态(插图 1a-d中的照片)。

CO 氧化是一种常用的探针反应,用于评估基于 Pt-CeO2的催化剂和许多其他催化剂体系的催化氧化性能。反过来,Pt-CeO2 催化剂局部结构的差异也可以通过其相应的 CO 氧化活性反映出来。如图 1e 所示,随着煅烧温度的升高,Pt/CeO2催化剂上的 CO 氧化活性单调下降。Pt/CeO2-350 (图 1a)上铂团簇的存在很好地解释了其最佳 CO 氧化活性,因为铂团簇-CeO2 界面位点被认为是 CO 氧化最活跃的物种。虽然 Pt/CeO2-550、Pt/CeO2-700 和 Pt/CeO2-800 上的铂物种都是铂单原子形式,但 Pt/CeO2-550、Pt/CeO2-700 和 Pt/CeO2-800 上 CO 氧化活性的显著差异表明,在这三种催化剂上成功产生了不同状态的铂单原子。
H2 -TPR技术是评估Pt物质的分散性以及Pt/CeO2基催化剂上Pt-O-Ce相互作用强度的有力工具5。在此,收集了Pt/CeO2 -550、Pt/CeO2 -700、Pt/CeO2-800的H2-TPR曲线,以研究Pt单原子与CeO2载体之间的相互作用(图 2a,b) 。 H2消耗峰值集中在 150 °C 左右,这可能是由于 Pt-O-Ce 结构的还原,而 H2消耗峰值位于大约 420 和785 °C 可以分别归因于表面Ce4+物质和本体CeO2的减少6。有趣的是,随着煅烧温度从 550 °C 升高到 800 °C,归因于 Pt-O-Ce 结构还原的H2消耗峰转移到更高的温度(126 °C → 173 °C)且强度增强,表明Pt-O-Ce相互作用的不同强度以及Pt单原子在Pt/CeO2-X催化剂上的不同位置。对于Pt/CeO2 -700,Pt单原子应处于Pt/CeO2-550和Pt/CeO2-800中Pt物种的混合状态。基于CO氧化评价结果,Pt/CeO2 -800催化剂表现出与纯CeO 2载体非常相似的CO氧化活性。因此,Pt/CeO2-800 上较高还原温度下 Pt-O-Ce 物质的 H2消耗峰更加强烈,表明该催化剂上的 Pt 单原子可能已经迁移到 CeO2载体的表面晶格中,并且孤立的Pt原子通过更多的Pt-O-Ce键与CeO2牢固结合。
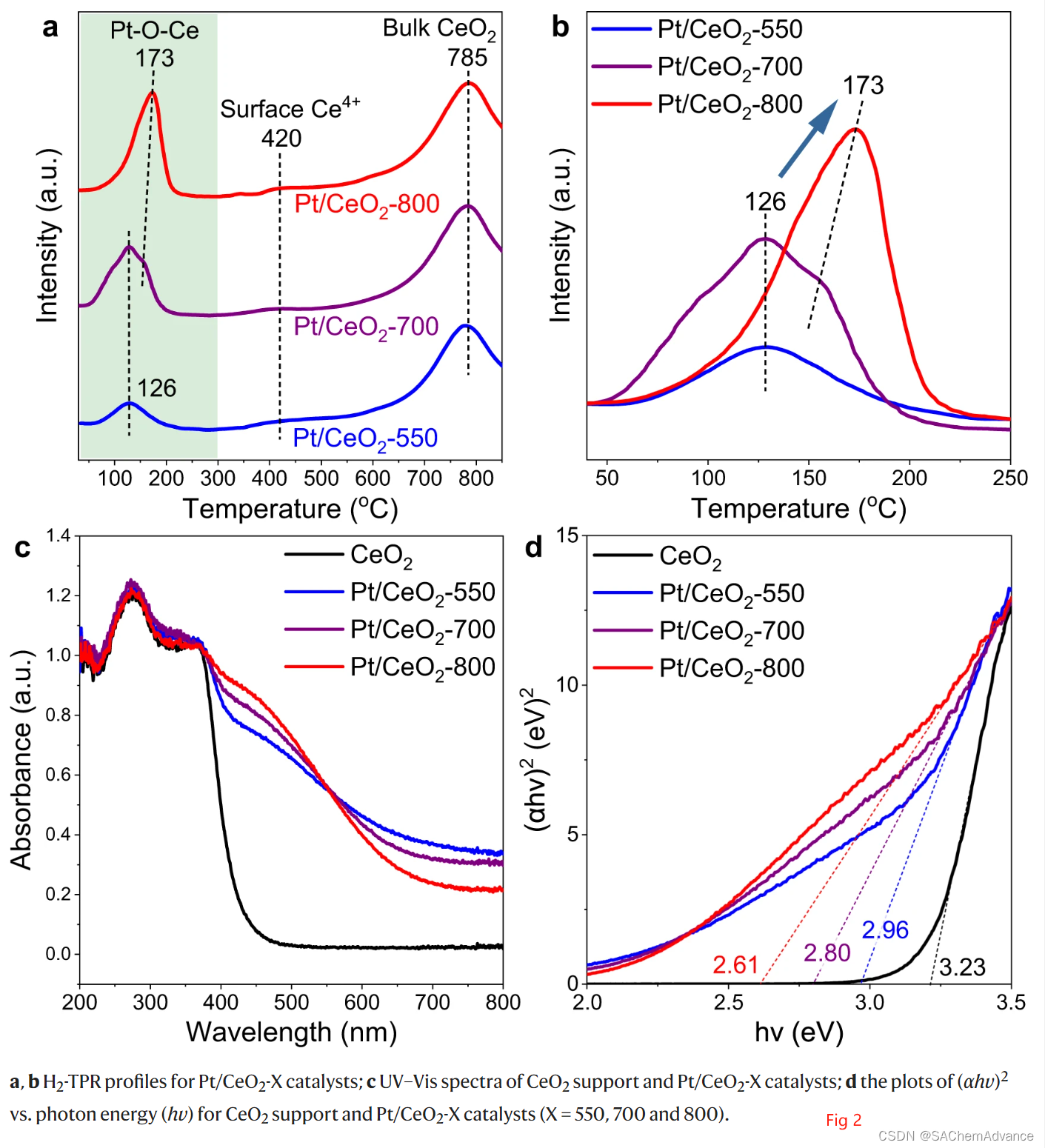
为了研究铂单原子与 CeO2 载体之间的相互作用,还进行了紫外可见光谱分析。如图 2c 所示,CeO2和 Pt/CeO2-X 催化剂(X = 550、700 和 800)上 400 nm 以下的波段可归因于 O2p → Ce4f 转变的电荷转移。有趣的是发现浸渍Pt后,在 400-600 纳米波长处观察到了额外的强吸收,被归类为absorption tail或 Urbach tail ,这与缺陷累积引起的带隙涂抹有关7。对于 Pt/CeO2-X 催化剂,这种吸收尾可能是由于高度分散的铂物种引起的结构紊乱。此外,随着煅烧温度的升高,400-600 nm 处的吸收强度也相应增强,这表明在更高温度下煅烧的 Pt/CeO2 催化剂具有更高的表面结构紊乱和更多的表面缺陷,这可能是由于铂单原子迁移到 CeO2 晶格中以及铂与 CeO2 载体之间更强的相互作用所致。甲醇吸附实验的原位 DRIFTS(可用来表征缺陷)进一步证实了 Pt/CeO2-800 上形成了更多的表面缺陷,其中 Pt/CeO2-800 上 1034 cm-1 处的条带强度远高于原始 CeO2和 Pt/CeO2-550上的谱带强度。为了进一步了解 Pt/CeO2-X 催化剂中铂物种与 CeO2 载体之间的相互作用,还利用 Davis 和 Mott 方程计算了间接带隙(图 2d 和表 1)。Pt/CeO2 -X 催化剂的带隙远低于 CeO2 载体的带隙(3.23 eV),这表明金属水平插入了 CeO2 的价带和导带之间,并且通过形成 Pt-O-Ce 键产生了强烈的相互作用。Pt/CeO2-800 的带隙(2.61 eV)低于 Pt/CeO2-550 (2.96 eV)和 Pt/CeO2-700 (2.80 eV),这充分证明了在 Pt/CeO2-800 催化剂上形成了更强的 Pt-O-Ce 相互作用的观点。
Pt单原子的位置和配位环境(Location and coordination environment of Pt single atoms)
通过 Pt/CeO2-550 和 Pt/CeO2-800 的 AC-HAADF-STEM 图像,以确定铂物种的位置(图 3)以及 CeO2载体选择性地暴露出 (110) 晶面。不出所料,在 Pt/CeO2-550 和 Pt/CeO2-800 上没有发现可识别的铂簇,只能观察到孤立铂原子的亮点。高分辨率 EDS 图谱结果还表明,铂物种在 Pt/CeO2-550 和 Pt/CeO2-800 上都处于高度分散状态(补充图 3)。此外,还发现这两种催化剂上分离出的铂单原子与 CeO2(110)平面上的 Ce 柱非常吻合。根据原位 DRIFTS 对 CO 的吸附、CO 氧化活性评估、H2-TPR 和紫外可见光谱的结果,可以推断出 Pt/CeO2-550 和 Pt/CeO2-800 上的铂单原子应处于不同的状态,包括位置和配位环境。考虑到AC-HAADF-STEM图像只是投影图,Pt/CeO2-550和Pt/CeO2-800上的Pt单原子可能位于CeO2载体上Ce的不同取代位点或外延生长位点,这使得Pt单原子处于不同的状态,但都在CeO2(110)平面上的Ce柱中观察到。借助 AC-HAADF-STEM 图像的线轮廓(图 3a、b),可以很好地确定Pt/CeO2-550 和 Pt/CeO2-800 上 Pt 单原子的不同位置。看出了。对于Pt/CeO2-550,Pt原子主要位于CeO2表面的边缘或台阶位点,而Pt/CeO2-800上的Pt原子可能位于CeO2台阶上的Ce取代位点(例如,嵌入CeO2的表面晶格中)。
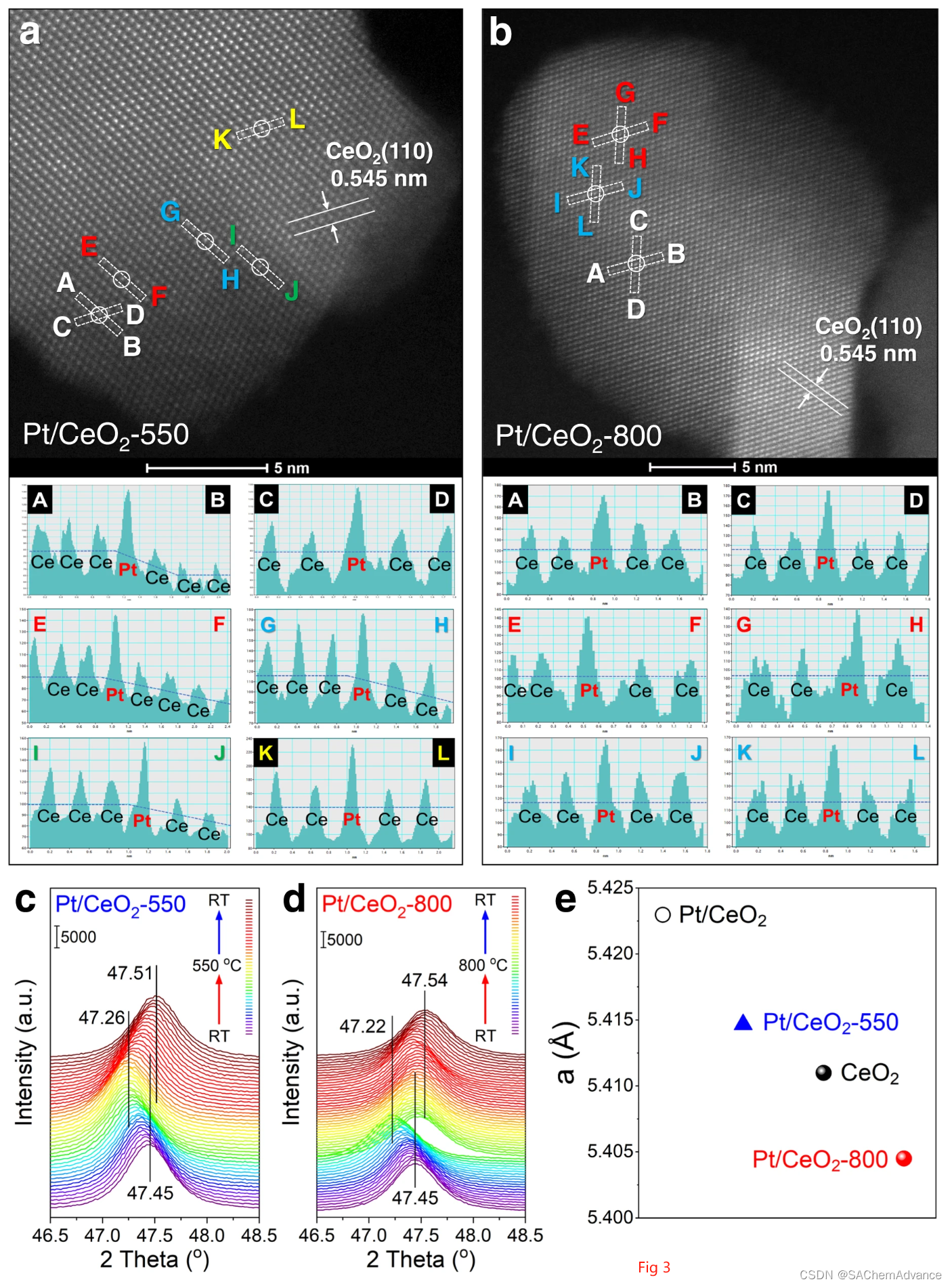
为了进一步揭示Pt原子在CeO2载体上的位置,设计了两组原位XRD实验,其中在整个样品加热(至550或800°C)和冷却过程中连续收集XRD图谱(补充图1)。 5和图 3c、d )。测量前,将新浸渍Pt硝酸溶液的CeO2粉末在空气中于120℃干燥30分钟以去除过量的H2O(表示为Pt/CeO2)。在煅烧获得Pt/CeO2-550的过程中,从室温到550℃的加热阶段,XRD峰向较低角度偏移,在冷却阶段又向较高角度偏移,这应该与晶格膨胀和晶格收缩有关8。在获得Pt/CeO2-800的煅烧过程中,由于CeO2的晶格膨胀,在加热阶段(<620℃),XRD峰也向较低角度移动。然而,在约 620 °C 的高温下,Pt/CeO2-800 的 XRD 峰突然转回高角度,这应该是由于在高温下 O2解吸形成了表面 Ce2O3 物种9。空白CeO2载体的XRD峰在加热和冷却阶段显示出与Pt/CeO2-550和Pt/CeO2-800催化剂相似的移动趋势(补充图 6)。在550和800℃焙烧后,Pt/CeO2催化剂的XRD衍射峰不同程度地向更高角度偏移。考虑到CeO2载体已经通过在800℃下煅烧12小时而预稳定,Pt/CeO2衍射峰的移动应该是由于Pt物质引起的CeO2晶格膨胀或收缩10。通过对Pt/CeO2、Pt/CeO2-550和Pt/CeO2-800的XRD图谱进行Rietveld精修,计算了这些样品上 CeO2的精修晶格参数(图3e)。值得注意的是,未煅烧的Pt/CeO2和Pt/CeO2-550的晶格参数略高于标准CeO2参考,这应该是铂沉积在 CeO2表面的扰动效应所致。有趣的是,当煅烧温度升高到800℃时,观察到晶格参数急剧下降,这可能与离子半径较小的Pt离子(Ce4+= 0.970 Å,Ce3+ = 1.280 Å,Pt4+ = 0.625 Å,Pt2+ = 0.800 Å)并进一步畸变有关,这与之前的报道一致,即在高温煅烧过程中,Pt2+可能会扩散到 CeO2 晶格中。原位 XRD 结果进一步表明,在 Pt/CeO2-800 催化剂中,铂原子可能已融入 CeO2 梯级的表面晶格中。
XAS(X射线吸收光谱)进一步阐明了 Pt/CeO2-X 催化剂上铂单原子的价态和配位环境,如图 4a 所示,Pt/CeO2 催化剂上 Pt-L3 XANES 的白线强度(近边区域的强吸收)始终远高于铂箔的白线强度,但低于 PtO2 的白线峰强度,这表明 Pt/CeO2-X 催化剂上的铂物种处于 0 和 +4 之间的中间价态。有趣的是,随着煅烧温度的升高,Pt/CeO2 催化剂上 Pt-L3 XANES 的白线强度单调下降,表明铂价态相应降低。通过 XANES 线性组合拟合进一步确定了铂物种的平均价态。正如预期的那样,Pt/CeO2-800 上的铂物种确实比 Pt/CeO2-550 上的铂物种(3.4 ± 0.1)显示出更低的价态(2.7 ± 0.1),这也得到了铂 4f XPS 结果的进一步支持,即与 Pt/CeO2-550 上的铂物种相比,Pt/CeO2-800 上产生了更多的 Pt2+ 物种。
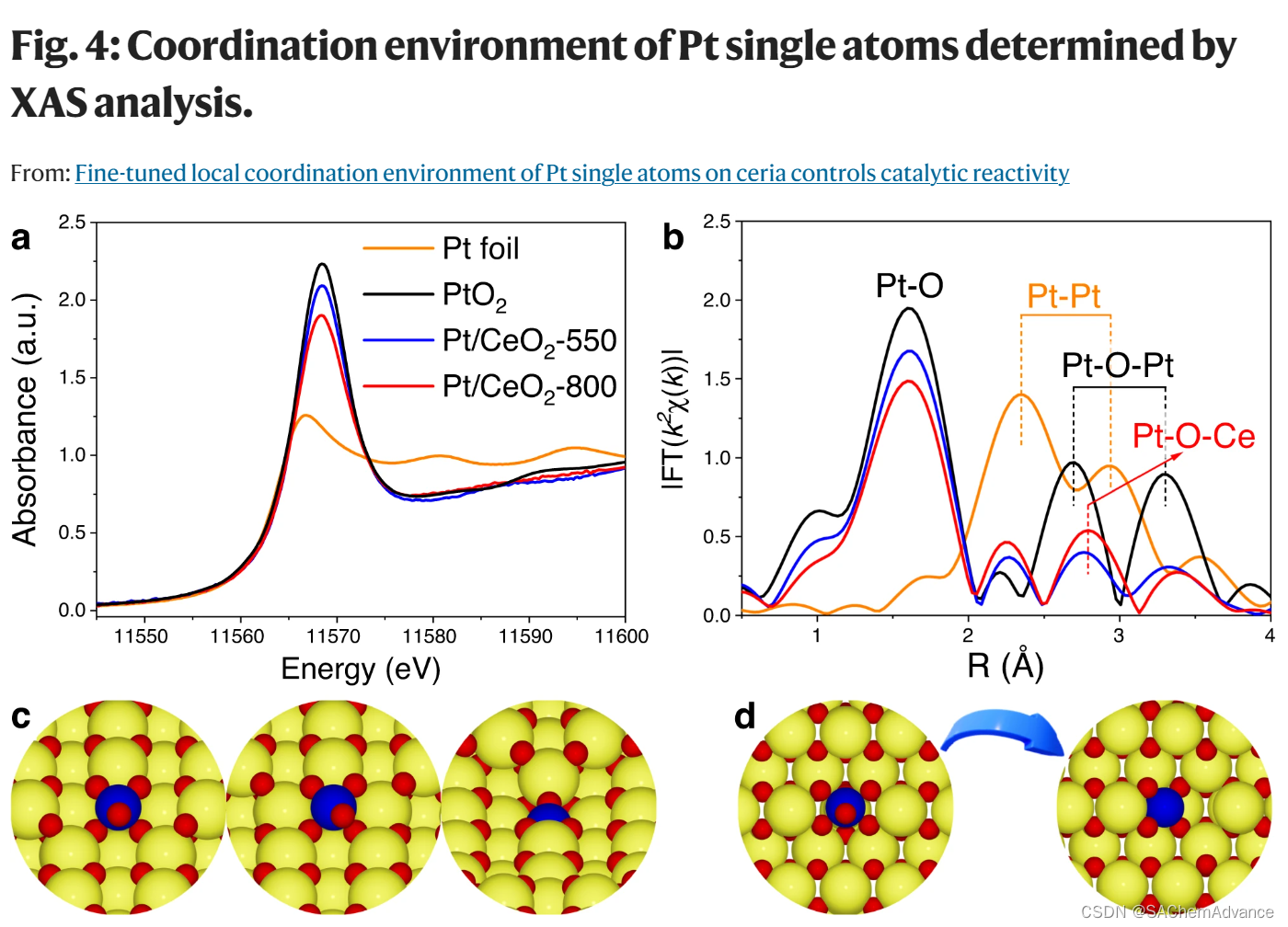
在此,为了进一步揭示Pt单原子在Pt/CeO2-550和Pt/CeO2-800催化剂上的局域配位结构,进行了EXAFS曲线拟合,并将结果绘制在R空间和k空间中(图2)。 (图4b、补充图 7b、补充图 10和补充表 3 )。 Pt-O-Pt和Pt-Pt配位层的缺失以及Pt-O和Pt-O-Ce配位层的独特观察进一步证明了Pt/CeO2-550和Pt/CeO2-800上Pt单原子的形成。尽管 Pt/CeO 2 -800 (CNPt– O = 4.4 ± 0.3 ) 确实表现出 比 Pt/CeO2-550 (CNPt– O = 5.1 ± 0.4)更低的 Pt–O 配位数 (CN) ,但与 Pt/CeO2-550 (CNPt– O– Ce = 3.7 ± 0.3) 上观察到的相比,在 Pt/CeO2-800(CNPt– O– Ce = 4.2 ± 0.3 ) 上观察到 Pt–O–Ce 的更高的配位数 ,进一步验证了Pt/CeO2-800内更多Pt-O-Ce键的形成,这可以通过拉曼光谱进一步证明。如上所述,对于Pt/CeO2-550,Pt单原子应主要位于CeO2(110)的边缘或台阶位置,这不会引起CeO2的任何晶格收缩(图 4c )。
相反,对于Pt/CeO2-800,CeO2载体晶格的显著收缩可能与Pt原子掺杂进入CeO2(110)的表面晶格有关。在理想模型中,对于Pt原子取代CeO2(110)上表面Ce位点,CNPt– O应为6,高于Pt/CeO2-800上的CNPt– O(CNPt– O= 4.4±0.3)。然而,由于 Pt2+物种具有比 Ce4+(约0.970 Å) 更小的离子半径 (约0.800 Å),因此 Pt 单原子从六配位位点到方形平面配位位点的显着重建可能发生在Pt/CeO2-800来平衡空间和电荷,从而导致CNPt– O从6 减少到4(图4d)。据报道,Pt 单原子的类似重构也发生在 CeO2(111) 上11。
Pt 1配位环境的DFT计算
为了证实关于铂单原子在 Pt/CeO2-550 和 Pt/CeO2-800 上的位置和配位环境的猜想,进行了 DFT 理论计算。对于 Pt/CeO2-550,根据实验证据表明铂单原子主要位于 CeO2(110)表面的边缘/台阶位点或其附近,提出了十种构型(图 5a1-a10)。
其中,用一个铂原子取代边缘位点上的一个铈原子构建了五种构型(图 5a1-a5)。在 Pt@CeEdge 构型中,铂取代了边缘位点的 Ce 原子,并与 6 个氧原子配位形成八面体 Pt-O6(图 5a1)。一旦 Pt@CeEdge 上的一个或两个 O 原子被移除,Pt@CeEdge 构型就演变成 Pt@CeEdge-O 或 Pt@CeEdge-2O(图 5a2-a3),其中铂分别与 5 或 4 个氧原子配位形成 Pt-O5 或方形平面 Pt-O4。进一步去除另一个氧原子后,形成了 Pt@CeEdgeOv(1) (图 5a4)或 Pt@CeEdgeOv(2)(图 5a),其配位为 Pt-O3。
此外,还构建了另外五种铂原子位于阶跃位点或阶跃位点附近的构型。在 Pt@Step 构型中,铂原子以 Pt-O4 配位吸附在 CeO2(110)表面的阶梯上(图 5a6)。与 Pt@CeEdge 构型类似(如图 5a7-a9 所示),在添加或去除 O 原子后,得到了三种新的构型(Pt@Step+O、Pt@StepOv (1) 和 Pt@StepOv (2))。此外,还提出了一种Pt取代 Ce 阶梯原子的构型(Pt-O4 配位)(图 5a10)。

此处省去了CO反应以及NH3氧化反应对应内容
讨论
通过简单的煅烧温度控制策略,在 CeO2载体上精确制备了具有不同局部配位环境的铂单原子催化剂。随着煅烧温度从 350 ℃升至 800 ℃,铂团簇首先分散为铂单原子,然后通过 Pt-O-Ce 连接与 CeO2 形成相互作用更强的铂单原子。与 Pt/CeO2-550 催化剂中的铂单原子主要位于 CeO2载体的边缘位点不同,在 Pt/CeO2-800 催化剂中,铂单原子主要融入 CeO2 的表面晶格中,并通过进一步的结构重构在 CeO2梯面上形成类似方形平面的配位环境。虽然 Pt/CeO2-800 催化剂上的铂单原子在 CO 氧化反应中的催化活性因其活化 O2 的能力较弱而受到限制,但由于其表面酸性增强,有利于 NH3 的吸附/活化,且反应过程中产生的 H2O 更容易解吸,因此 Pt/CeO2-800 的 NH3 氧化活性远高于 Pt/CeO2-550 催化剂。这表明,精确控制铂单原子的配位环境有利于最大限度地提高它们在不同氧化反应中的催化性能。这项研究为灵活调节单原子催化剂的局部配位环境,使其更有效地应用于不同的目标反应提供了启发。
方法
催化剂制备
载体前处理:对商用 CeO2(高比表面积铈,BET 表面积约为 120 m2/g)在 800 °C 的空气中预煅烧 12 小时,以尽量减少其在后续催化剂制备过程中可能发生的结构变化。为了制备 CeO2支持的铂催化剂,以 Pt(NO3)2 为前驱体,通过初湿浸渍 (IWI) 法将 1.00 wt.% 的铂负载到预处理过的 CeO2 载体上。将 Pt(NO3 )2 浸渍到 CeO2 上后,将湿粉末在 120 ℃ 的空气中干燥 1 小时,然后在 350、550、700 或 800 ℃ 的空气中煅烧 2 小时。通过电感耦合等离子体-光发射光谱(ICP-OES)测定了所选催化剂中铂的实际质量负载。不出所料,Pt/CeO2 -550(0.92 wt.%)和 Pt/CeO2 -800(0.90 wt.%)的铂负载量几乎相同,非常接近标称铂负载量(1.00 wt.%)。
表征(部分可参考)
- 物理吸附:The specific surface area and pore volume were measured by N2 physisorption at −196 °C using a Micromeritics ASAP-2020 analyzer. Before each test, the samples were degassed at 300 °C for 4 h. The specific surface area was calculated using Brunauer-Emmett-Teller method, and the pore volume and pore size distribution were determined by BJH method using the desorption isotherms.
- 原位红外:In situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) study was carried out on a Thermo Nicolet iS50 FTIR spectrometer equipped with a liquid nitrogen-cooled mercury–cadmium–telluride (MCT) detector. In each test, 50 mg of catalyst powder was placed in the DRIFTS cell (PIKE Technologies DiffusIR), pressed, and mounted. The catalysts were first pretreated in air at 300 °C for 1 h. Afterwards, the catalysts were cooled down step-wise to room temperature. During this process, the background spectra were collected at target temperatures for different experiments (e.g., 25 °C for CO adsorption, 25 °C for methanol adsorption, 100/125/150/175/200/225/250 °C for NH3 adsorption/desorption, and 175 °C for NH3 oxidation). For CO adsorption, methanol adsorption, or NH3 adsorption/desorption/oxidation experiments, the feed gas consisted of 1% CO, 15% methanol, or 500 ppm NH3 and/or 5 vol% O2 (when used), respectively, using Ar as balance. The total flow rate was controlled at 83.33 mL∙min−1. All the spectra were collected from 400 to 4000 cm−1at a resolution of 4 cm−1 for 100 scans. 甲醇吸附可以验证缺陷量
- H2-TPR:H2-temperature-programmed reduction (H2 -TPR) was conducted on a Quantachrome Autosorb-iQ instrument. Typically, 30 mg of sample was loaded in a U-type quartz tube reactor and pretreated with air at 400 °C for 1 h. When the sample was cooled down to room temperature in air, the feeding gas was switched to 10 vol% H2 balanced with Ar (30 mL∙min−1). Then, the reactor was heated linearly to 900 °C with a ramping rate of 10 °C∙min−1. The signal of H2 consumption was monitored by a thermal conductivity detector (TCD). H2 O moisture in the gas mixture was removed by a cold trap filled with liquid N2 before passing into the TCD.
-紫外可见光谱:Ultraviolet-visible (UV–Vis) spectra were collected on a Shimadzu UV-2401 PC instrument in diffuse mode. BaSO4 was used as a reference and the spectra collection range was 200−800 nm. Samples were exposed to air at room temperature throughout the experiments. - AC-TEM:Atomic-resolution aberration-corrected high angle annular dark field scanning transmission electron microscopic (AC-HAADF-STEM) images were collected on a FEI Titan Cubed G2 60-300 aberration-corrected S/TEM instrument at 300 kV accelerating voltage. The observations were performed in the HAADF mode, which allowed Z-contrast imaging. The probe convergence angle on the Titan electron microscope was 22 mrad, and the angular range of the HAADF detector was from 79.5 to 200 mrad. To prepare the TEM samples, an appropriate amount of sample powder was dispersed in ethanol and then dropped on a 3 mm TEM Mo grid. The images of energy dispersive spectroscopy (EDS) elemental mapping in the STEM mode were obtained from the Titan electron microscope using SuperX system.
- XPS: X-ray photoelectron spectroscopy (XPS) was collected on a VG CLAM 4 MCD analyzer. Before the analysis, the samples were degassed in a preparation chamber (10−5 Torr) for 0.5 h. Then, the samples were put into the analysis chamber (3 × 10−9 Torr) for further analysis. The binding energies (BE) were calibrated with C1s line at 284.6 eV.
- 拉曼:Raman spectra were collected on a Renishaw Laser Raman spectrometer (Renishanplc) with an Ar+ laser beam. The emission line was 532 nm and the output power was 10 mW. Samples were exposed to air at room temperature throughout the experiments.
- NH3-TPD: NH3-temperature-programmed desorption (NH3-TPD) was carried out in a fixed-bed quartz flow reactor connected with an online Thermo Nicolet iS10 FTIR spectrometer equipped with a 2 m path-length gas cell (200 mL volume). In each test, 100 mg sample was pretreated by pure Ar at 200 °C for 1 h and then saturated with NH3 at 100 °C. Then, Ar was introduced into the quartz tube reactor again to remove the weakly adsorbed NH3. Finally, the sample was heated linearly to 500 °C at a rate of 10 °C∙min-1 in the flowing Ar (100 mL∙min-1).
Fine-tuned local coordination environment of Pt single atoms on ceria controls catalytic reactivity
声明:仅代表推文作者个人观点,作者水平有限,如有不科学之处,请大家指正!
Ma, Y. et al. Tailoring of the proximity of platinum single atoms on CeO2 using phosphorus boosts the hydrogenation activity. ACS Catal. 9, 8404–8412 (2019). ↩︎
Jeong, H. et al. Controlling the oxidation state of Pt single atoms for maximizing catalytic activity. Angew. Chem. Int. Ed. 59, 20691–20696 (2020). ↩︎
Jiang, D. et al. Tailoring the local environment of platinum in single-atom Pt1/CeO2 catalysts for robust low-temperature CO oxidation. Angew. Chem. Int. Ed. 60, 26054–26062 (2021). ↩︎
Wan, W. et al. Highly stable and reactive platinum single atoms on oxygen plasma-functionalized CeO2 surfaces: Nanostructuring and peroxo effects. Angew. Chem. Int. Ed. 61, e202112640 (2022). ↩︎
Gao, Y., Wang, W., Chang, S. & Huang, W. Morphology effect of CeO2 support in the preparation, metal-support interaction, and catalytic performance of Pt/CeO2 catalysts. Chem. Cat. Chem. 5, 3610–3620 (2013). ↩︎
Tan, W. et al. Tuning single-atom Pt1-CeO2 catalyst for efficient CO and C3H6oxidation: Size effect of ceria on Pt structural evolution. ChemNanoMat 6, 1797–1805 (2020). ↩︎
Bugrova, T. A. et al. Insights into formation of Pt species in Pt/CeO2 catalysts: Effect of treatment conditions and metal-support interaction. Catal. Today 375, 36–47 (2021).Return to ref 43 in article ↩︎
Parvulescu, V. I. & Tiseanu, C. Local structure in CeO2 and CeO2-ZrO2 nanoparticles probed by Eu luminescence. Catal. Today 253, 33–39 (2015). ↩︎
Ma, R., Jahurul Islam, M., Amaranatha Reddy, D. & Kim, T. K. Transformation of CeO2 into a mixed phase CeO2/Ce2O3 nanohybrid by liquid phase pulsed laser ablation for enhanced photocatalytic activity through Z-scheme pattern. Ceram. Int. 42, 18495–18502 (2016). ↩︎
D’Angelo, A. M., Webster, N. A. S. & Chaffee, A. L. Vacancy generation and oxygen uptake in Cu-doped Pr-CeO2 materials using neutron and in situ X-ray diffraction. Inorg. Chem. 55, 12595–12602 (2016). ↩︎
Su, Y.-Q. et al. Theoretical approach to predict the stability of supported single-atom catalysts. ACS Catal. 9, 3289–3297 (2019). ↩︎















 583
583











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









