







>ML建树原理
1.选择模型:首先需要根据不同的分子序列特点和研究目的,选择合适的进化模型。常用的模型包括JC模型、K80模型、HKY模型、GTR模型等。
2.构建初始树:为了进行ML优化,需要构建一颗初始的进化树。初始树的构建可以使用多种方法,例如随机构建、邻接法、UPGMA法等。
3.计算似然值:计算初始树的似然值,即给定当前进化树拓扑结构和分支长度下,观测到输入序列的概率。该概率可以根据所选的进化模型和序列数据计算得出。
4.优化树拓扑:通过修改进化树的拓扑结构,得到新的进化树,并计算其似然值。如果新的进化树的似然值比原始树更高,则接受该新树,否则舍弃。
5.优化分支长度:在确定树的拓扑结构后,进一步通过优化分支长度,使得观测到的序列数据出现的概率最大。这个过程通常使用牛顿法、梯度下降法等数值优化方法实现。
6.重复优化:不断重复步骤4和步骤5,直到ML似然值收敛或达到预设的停止条件。
7.输出结果:最终输出优化后的进化树和相应的似然值,用于进一步的分析和解释。
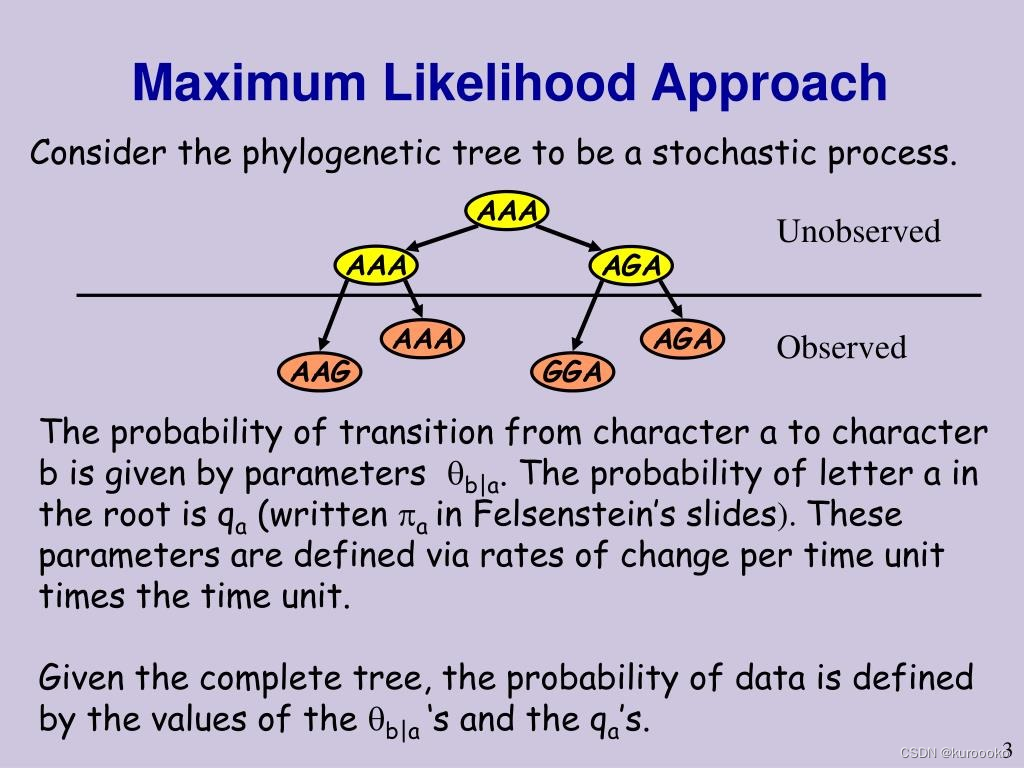
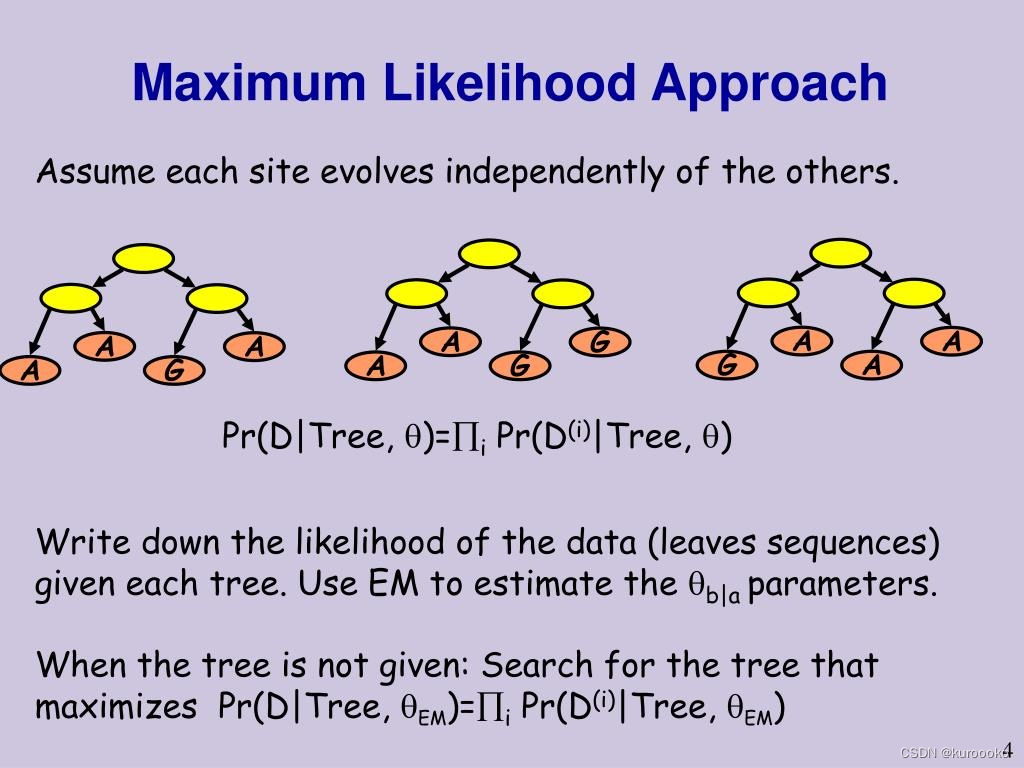
似然值是如何计算的:
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 2397
2397

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









