






一、TAD到底是否具有保守性
TAD结构的保守性的研究应该从两方面去看,是不同物种间的保守性,还是人类的不同组织或细胞系之间的保守性。目前还是存在一定的争论,因为最近在做TAD结构的研究,因此总结了一下最近看到的涉及TAD结构保守性的文章以及里面用到的TADs calling的方法。
二、方法总结
2.1 DI-HMM
文章标题:Topological domains in mammalian genomes identified by analysis of chromatin interactions
保守性:在人类中发现了76% 的小鼠边界,在小鼠中发现了54% 的人类边界;在人类胚胎干细胞和IMR90细胞中,胚胎干细胞中65%的边界在IMR90细胞系中发现,IMR90细胞中72%的边界在胚胎干细胞中发现;在人和小鼠中,组蛋白修饰标记的位点大约有60-75%的重叠。
分辨率:10kb-1Mb,边界识别分辨率40kb。
TADs calling方法:DI-HMM 通过识别基因组中相互作用频率的偏差,识别基因组中拓扑结构域和边界的位置。
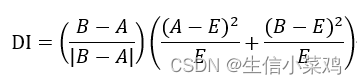
A为从给定的40kb bin映射到上游2Mb的read数,B是从相同的40kb bin映射到下游2Mb的read数,E是零假设下的期望read数,等于(A + B)/2。将方向性指数视为一种观测值,并认为“真正的”隐藏方向性偏差(DB)可以使用隐马尔可夫模型(HMM)来确定。HMM假设方向性指数观测值遵循高斯混合分布,然后预测状态为“上游偏差”、“下游偏差”或“无偏差”。 然后从整个基因组的HMM状态调用的结果推断出域和边界。域在单个下游偏置 HMM 状态的开头启动。该域在任何连续的下游偏态中都是连续的。然后,当到达一系列上游偏置状态中的最后一个时,域结束,域结束于最后一个HMM上游偏置状态的结束。
 保守性判断方法:计算两个细胞之间方向性指数的Spearman相关系数,在两种细胞类型中,如果隐马尔可夫模型(HMM)识别到某个边界,就会确定该边界的中心,并计算边界中心位置前后10个bin的方向性指数,然后在感兴趣的 两种细胞之间进行相关性分析。
保守性判断方法:计算两个细胞之间方向性指数的Spearman相关系数,在两种细胞类型中,如果隐马尔可夫模型(HMM)识别到某个边界,就会确定该边界的中心,并计算边界中心位置前后10个bin的方向性指数,然后在感兴趣的 两种细胞之间进行相关性分析。
为了进行随机相关性分析,研究者会从每种细胞类型中随机选择20个bin,然后计算这两种细胞类型之间这些随机选择的bin的Spearman相关系数。他们会重复这个随机选择的过程10,000次,以得到Spearman相关系数的随机分布。
最后,基于这些分析结果,如果某个边界区域在HMM调用中仅在一个细胞中被识别,并且在两种细胞类型之间的方向性指数没有明显的相关性,那么这个边界就会被称为“细胞类型特异性”。
边界大小:这些区域的拓扑边界小于400kb,76.33%的边界小于50kb。
2.2 Arrowhead箭头算法
文章标题:A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping
保守性:45%的小鼠结构域也存在于人类中
分辨率:在GM12878上以5kb分辨率执行箭头算法;以25 kb分辨率评估不同距离下域间接触与域内接触的比例;为了确定结构域结构如何影响染色质标记,将每个结构域分成10个bin,其中bin大小为结构域大小的十分之一;记录结构域边界左侧和右侧的10个bin中的平均染色质标记值时,bin大小设置为10kb的固定大小;探索保守性时以10kb的分辨率来识别域。
TADs calling方法:Arrowhead箭头算法 一个juicer工具,通过矩阵转换来揭示基因组中的特定结构。
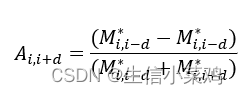
当且仅当轨迹i-d在定义域内,而轨迹i+d不在此范围内,![]() 是强阳的。当轨迹i+d在定义域内而轨迹i-d不在定义域内时,
是强阳的。当轨迹i+d在定义域内而轨迹i-d不在定义域内时,![]() 将是强负的。如果两个位点都在定义域内,或者两个位点都在定义域外,
将是强负的。如果两个位点都在定义域内,或者两个位点都在定义域外,![]() 将接近于零。
将接近于零。
计算![]() 的角点分数:
的角点分数:![]() La,b
La,b![]() 中条目的符号之和减去
中条目的符号之和减去![]() 中条目的符号之和;
中条目的符号之和;![]() 中条目的值之和减去
中条目的值之和减去![]() 中条目的值之和;
中条目的值之和;![]()
![]() 和
和![]() 的方差总和。通过计算每个可能的a,b的每个得分来对这三个子得分进行归一化,然后除以观察到的最大值。原始角点得分矩阵
的方差总和。通过计算每个可能的a,b的每个得分来对这三个子得分进行归一化,然后除以观察到的最大值。原始角点得分矩阵![]() 由所有像素
由所有像素![]() 的三个归一化得分之和组成。如果
的三个归一化得分之和组成。如果![]() 是真正的区域角落,
是真正的区域角落,![]() 的值通常会很大。
的值通常会很大。
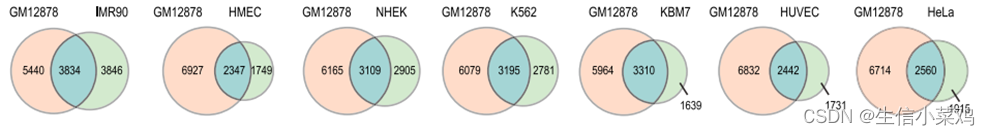
保守性判断方法:其他细胞系都与GM12878细胞系进行比较。如果在一个细胞类型中有一个以![]() 为右上角的被调用的区域,以及在GM12878中有一个以
为右上角的被调用的区域,以及在GM12878中有一个以![]() 为右上角的被调用的区域,满足
为右上角的被调用的区域,满足![]() 就认为是保守的。
就认为是保守的。
还通过检查GM12878结构域注释在所有其他细胞类型中的角点得分分布来评估结构域保守性。
边界大小:结构域边界左侧和右侧是个bin,bin大小为10kb
2.3 HiCExplorer
文章标题:TAD evolutionary and functional characterization reveals diversity in mammalian TAD boundary properties and function.
保守性:研究在不同物种之间TADs的保守性。发现14%的人类TAD边界在所有八个物种之间共享(超保守),而15%是人类特异性的。
分辨率:10kb
TADs calling方法:HiCExplorer工具中的hic-FindTADs. 该程序使用不同大小的运行窗口来测量每个Hi-C矩阵bin两侧之间的相互作用间隔或“绝缘分数”。“绝缘分数”通过使用滑动窗口计算接触的平均z-score来量化每个bin的相互作用。较小的绝缘分值表示较少的相互作用(较高的相互作用间隔)。TAD边界通过识别绝缘分数曲线中具有统计学意义的局部极小值来注释。
保守性判断方法:使用UCSC LiftOver工具将每个物种的TAD边界提升到人类基因组(hg38)坐标。没有提升的边界,或者提升到小于5kb区域的边界删除,使用bedtools工具将人类基因组坐标中彼此相距在10kb以内的所有边界合并。然后删除了合并后超过190kb的联合边界,将联合TAD边界与每个物种的提升边界相交,以确定跨物种边界的存在/不存在。
边界大小:原始边界宽度10kb,形成联合边界后大小不定。
2.4 TADLib(HiTAD)
文章标题:HiTAD: detecting the structural and functional hierarchies of topologically associating domains from chromatin interactions.
保守性:
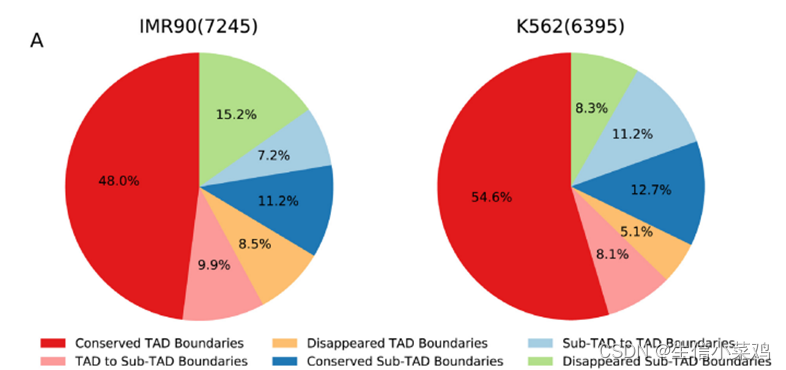
分辨率:20kb
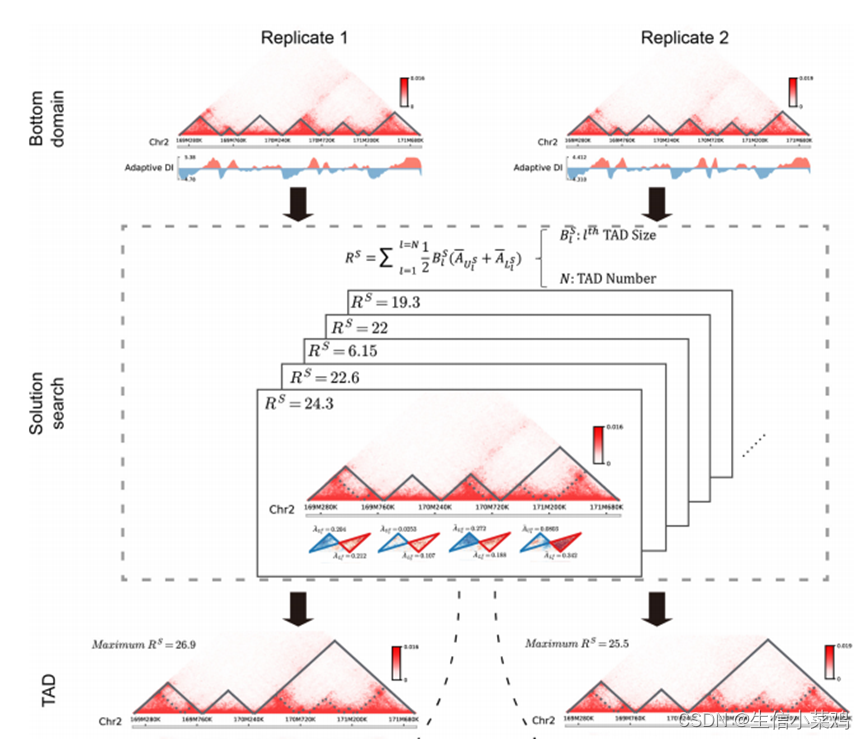
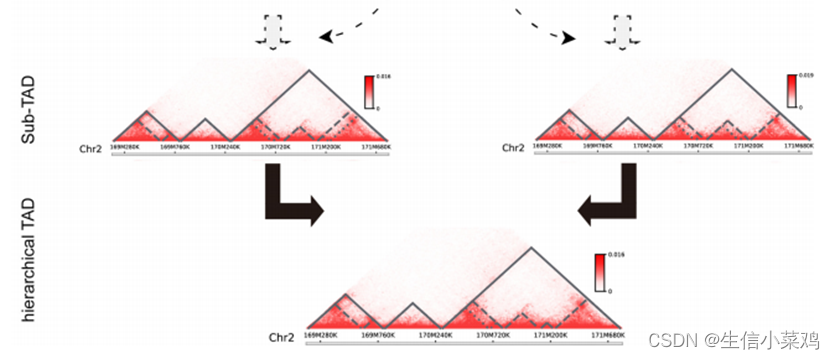
TADs calling方法:TADLib(HiTAD)结合TAD可以以分层的方式划分为更小的域这一事实,通过根据交互频率定义适当的目标函数,将分层TAD的检测转化为迭代优化过程。在这项工作中,目标函数被定义为域内相互作用频率和域间相互作用频率之间的富集,以减少基因组距离的影响。为了加快计算速度,提出了一种基于自适应方向性指数(DI)的隐马尔可夫模型(HMM),通过仅使用局部绝缘来敏感地生成全基因组的bottom domain。然后,在给定的目标函数下,利用全局染色体内相互作用,将这些bottom domain用作检测TAD的基本元件。为了更好地执行TAD检测,使用递归公式来解决优化问题。接下来,以类似的方式使用这些检测到的TAD来生成相应的子TAD,但将位于TAD内的底部域作为初始域。

















 2159
2159

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









