







参考我的上一篇博客https://blog.csdn.net/weixin_62528784/article/details/142236123?spm=1001.2014.3001.5502,
在处理完compartment模块之后,加下来要处理的就是TAD模块了,当然实际上这几个模块都是可以并行处理的,并没有严格的先后顺序,包括下一篇loop系列;
说回到TAD,传统的TAD分析可以说是有几种工具、有多少种分辨率就会有几种不同的结论,所以大多数hic数据分析过程中如非比亚并不会深入到TAD层面;
本人TAD分析中以hicexplorer为主,所以其对应TAD模块部分要熟悉:
以及GENOVA中TAD部分指南
1,首先是识别TAD:
此处使用hicexplorer的hicfindTAD函数来识别TAD,
具体细节参考官网https://hicexplorer.readthedocs.io/en/latest/content/tools/hicFindTADs.html
脚本其实很简单,类似PDX-HiC_processingScripts/8.TADs.sh,
脚本参考https://github.com/MaybeBio/TNBC-project/blob/main/3%2CTAD/TAD_find_merge.sh、https://github.com/MaybeBio/TNBC-project/blob/main/3%2CTAD/TADs.sh,
实际修改后为:HiC_pipeline/pipeline/hic 基础pipeline骨架/01_TAD_find_merge.sh,

本意上是call出了多个分辨率下的TAD,接下来想要将多个分辨率的数据合并起来,因为单个res下的TAD本身数据波动很大,不同工具以及不同res差异比较大,所以最开始的想法很简单,直接合并所有res下的数据,至少可信度高一点
(参考https://hicexplorer.readthedocs.io/en/latest/content/tools/hicMergeDomains.html,但是实际执行过程发现merge TAD会报错,所以不不建按照脚本中先call再merge操作,直接使用某一合适res进行call即可,本人选用40kb或20kb)
hicMergeDomains将来自hicFindTads的多个TAD域文件作为输入。它将不同分辨率的TAD合并到一个TAD域文件中,考虑已知TAD结合位点的蛋白峰,并计算出TAD的依赖图;
可以将各种分辨率的数据都call出来,然后使用merge domian将所有的TAD都合并起来;
不能用上全部数据也不能合并,建议还是使用单一res的数据;
事实上我们可以发现:
hicexplorer中也有merge loop的函数,可以考虑是否要在loop数据中合并多分辨率数据?
后1篇博客分析的时候我们发现实际上mustache中,原始脚本也在tsv层面上进行了loop结果的合并,不过那是后话了。
一些需要注意hicfindTAD参数的地方:
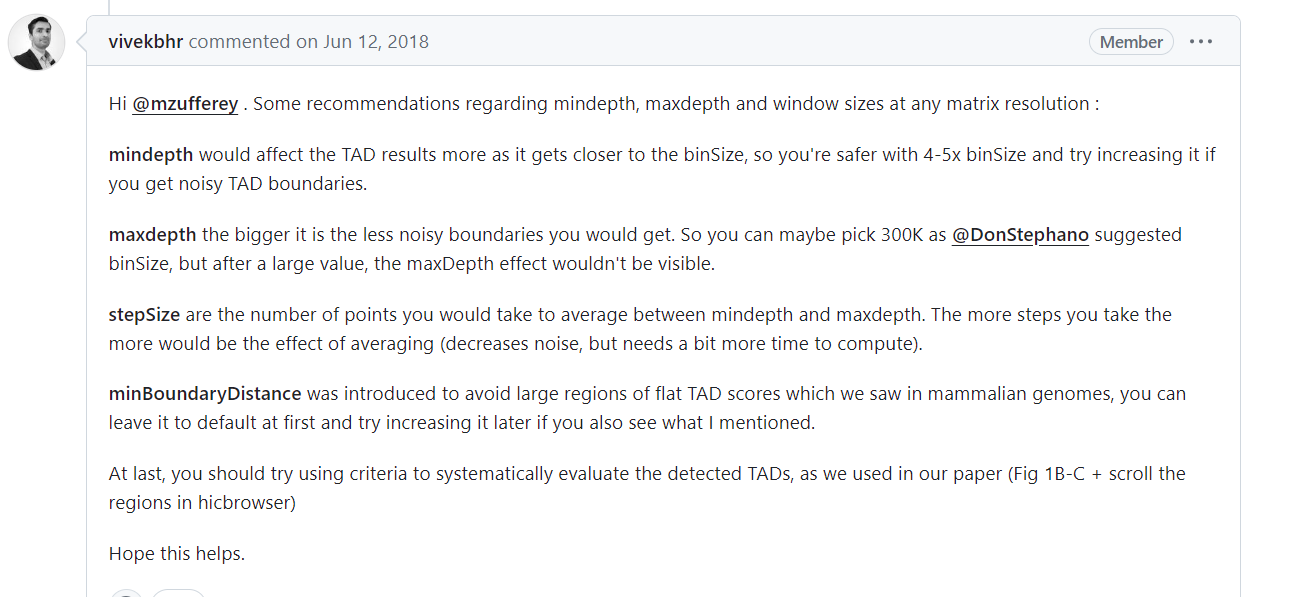
参考https://github.com/deeptools/HiCExplorer/issues/119
2,识别差异TAD并分析:
(1)识别差异TAD:
依旧是使用hicexplore中的函数hicDifferentialTAD
参考https://hicexplorer.readthedocs.io/en/latest/content/tools/hicDifferentialTAD.html

脚本参考https://github.com/MaybeBio/TNBC-project/blob/main/3%2CTAD/diff_TAD.sh,
即HiC_pipeline/pipeline/hic 基础pipeline骨架/02_diff_TAD.sh,
但是这里分析出来的差异TAD实际上绘图的效果与传统分析TAD边界来识别差异的方法是不一致的,
当时做的时候:很难绘制以及表述出来这种差异识别的原理,而且边界也没有那么容易处理(使用一维边界的信息来建模的话),肯定有bp差异,所以不能人工注释,必须使用工具;
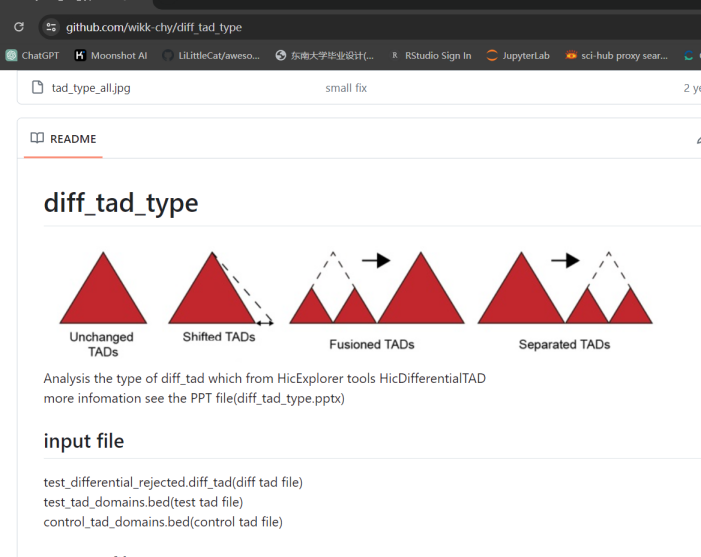
此处参考使用工具:https://github.com/wikk-chy/diff_tad_type
能够承接hicexplorer,且上游也是使用hicexplorer处理的
使用工具的执行脚本https://github.com/MaybeBio/TNBC-project/blob/main/3%2CTAD/diffTAD/type.sh,
即HiC_pipeline/pipeline/hic 基础pipeline骨架/03_diff_TAD_type.sh以及统计的04_stat_diffTAD.sh
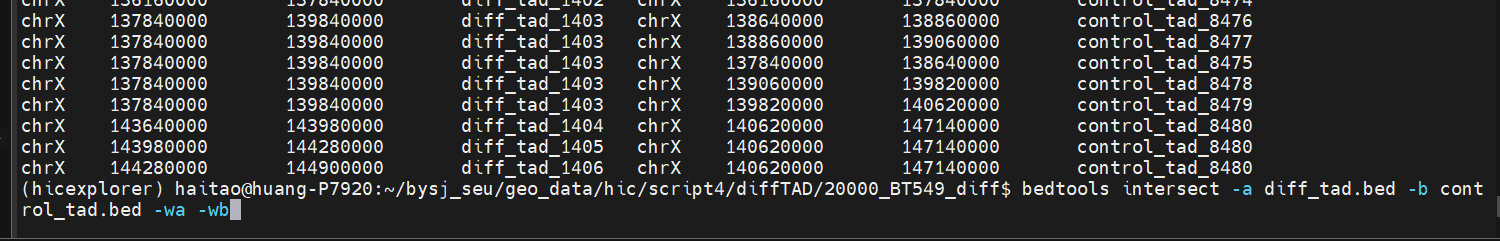
依据github中样本文件的格式,应该是问题出在bedtools上,
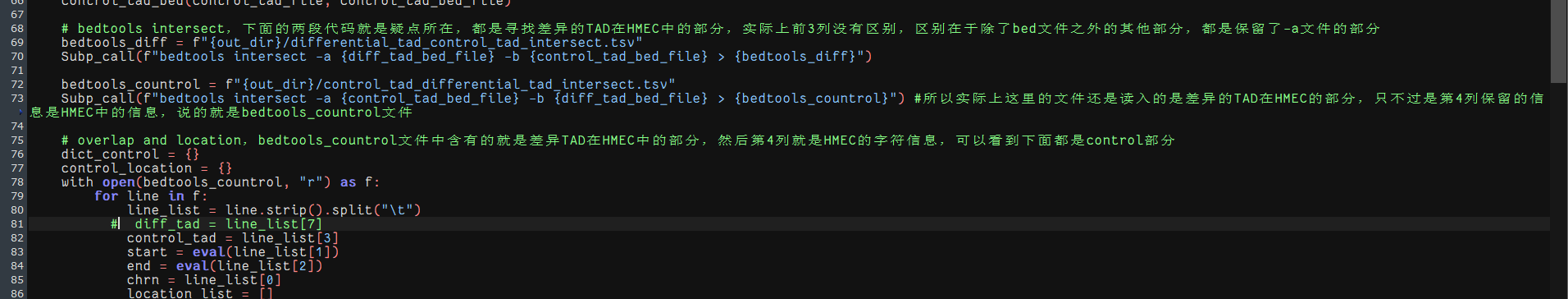
理论上来讲,应该是将处理之后的交集部分的TAD文件双方的信息都写入;
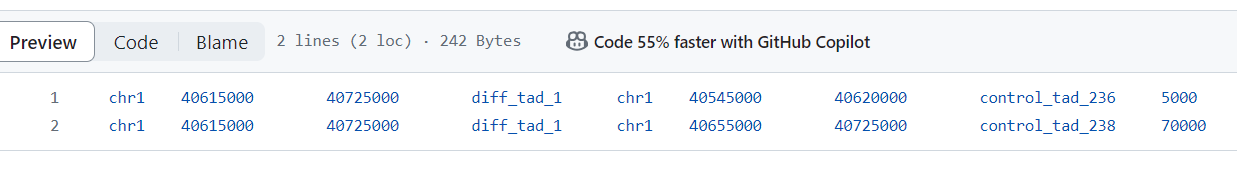

最后修改上只是在代码后面加了-wa,-wb部分
因为确实仅仅只是-a或-b的话,只能获取重叠区域——还是4列
代码中需要的是8列
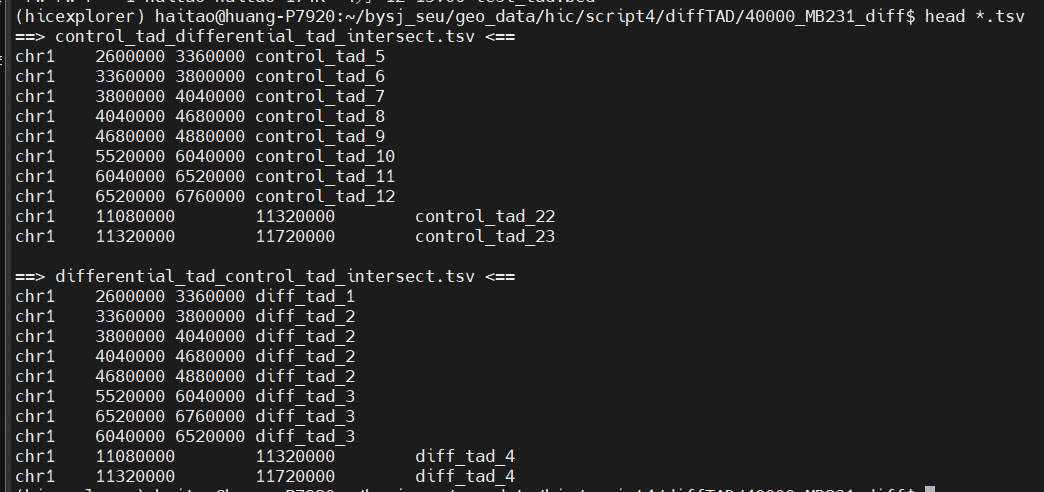
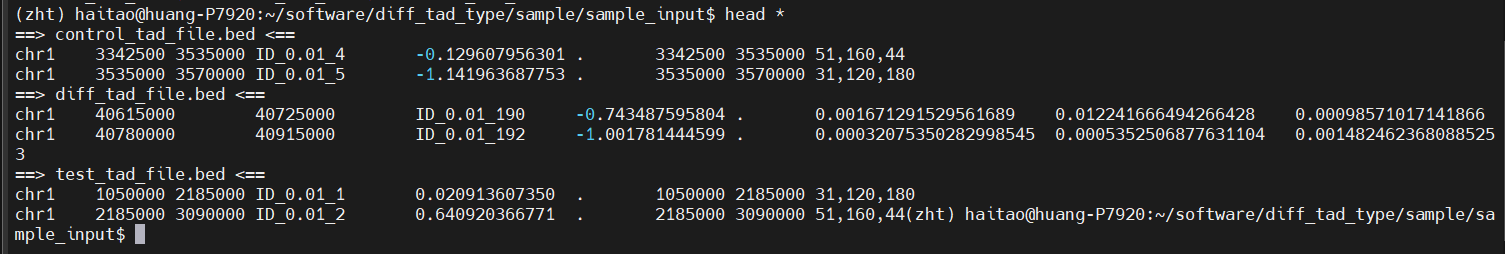
然后获取了这些文件:
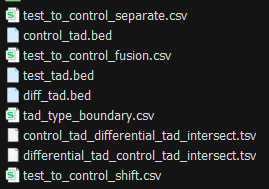
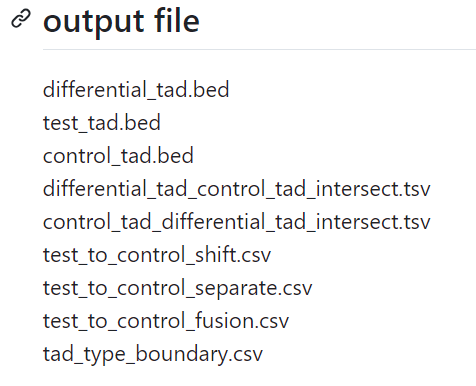
前3个是原始输入文件,中间是交集不用管
末尾4个文件才是重点
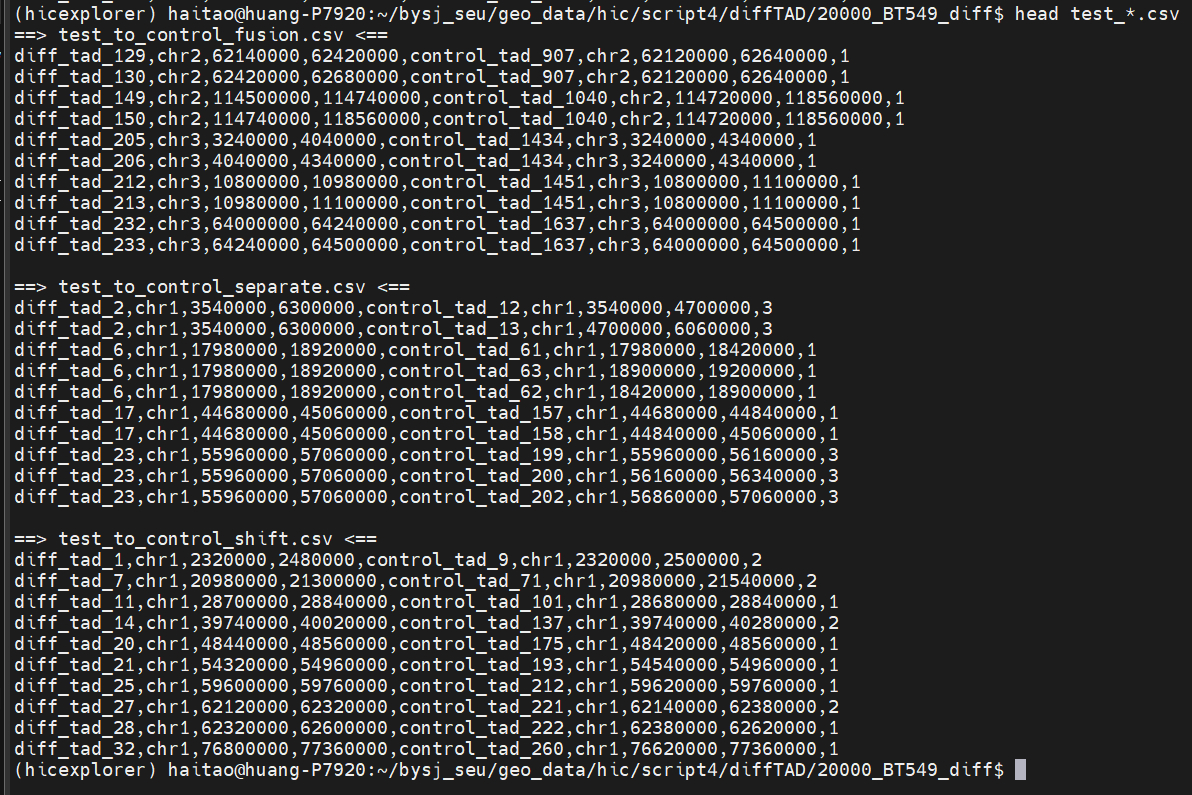
现在手头上有了这些变异类型的TAD的domain的bed信息,
比如说是:
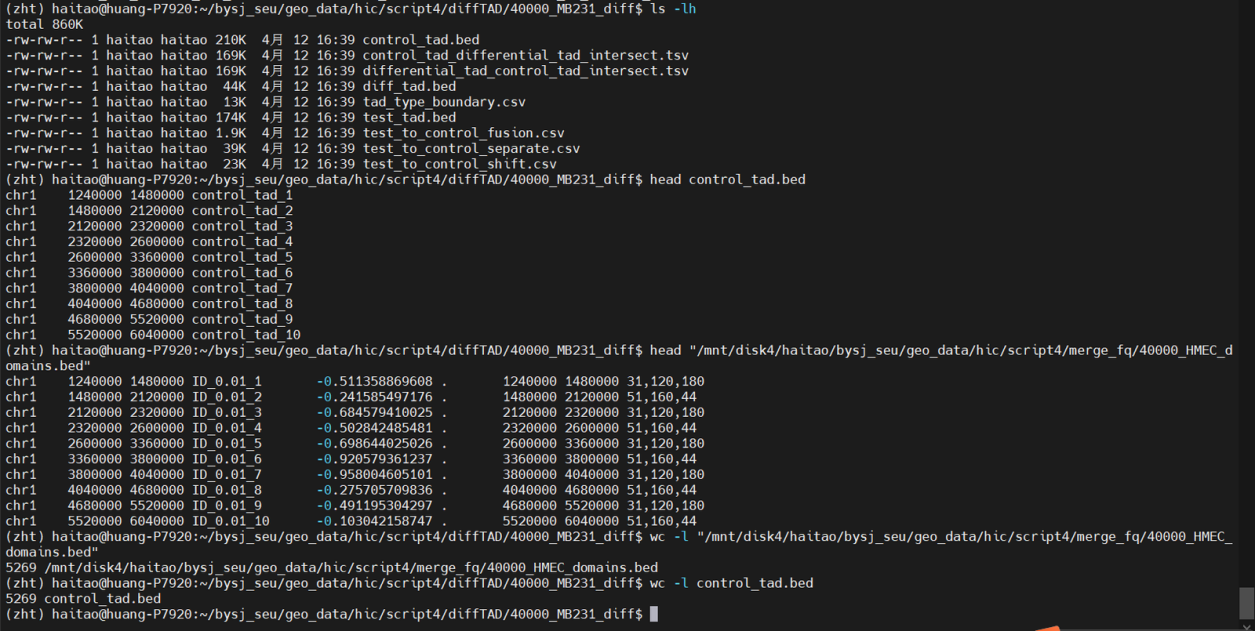
首先每个文件夹中control的tad都是HMEC的tad bed文件
test tad是对应TNBC细胞系的tad bed文件
diff是两者的差异tad的bed文件
然后具体diff中分shift,fusion,separate
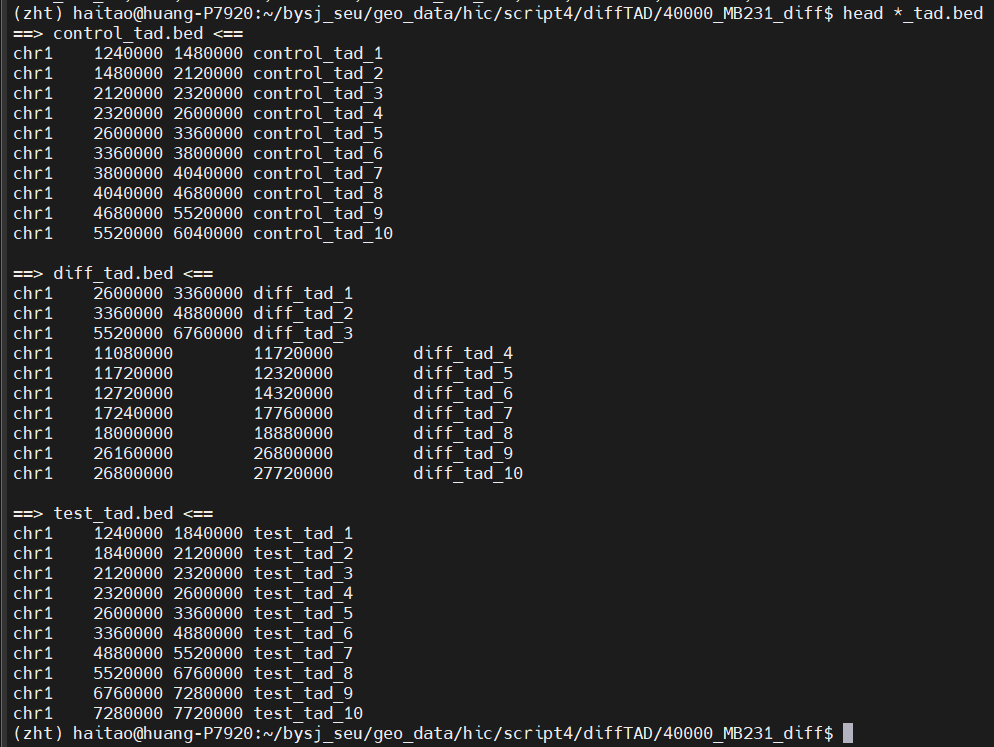
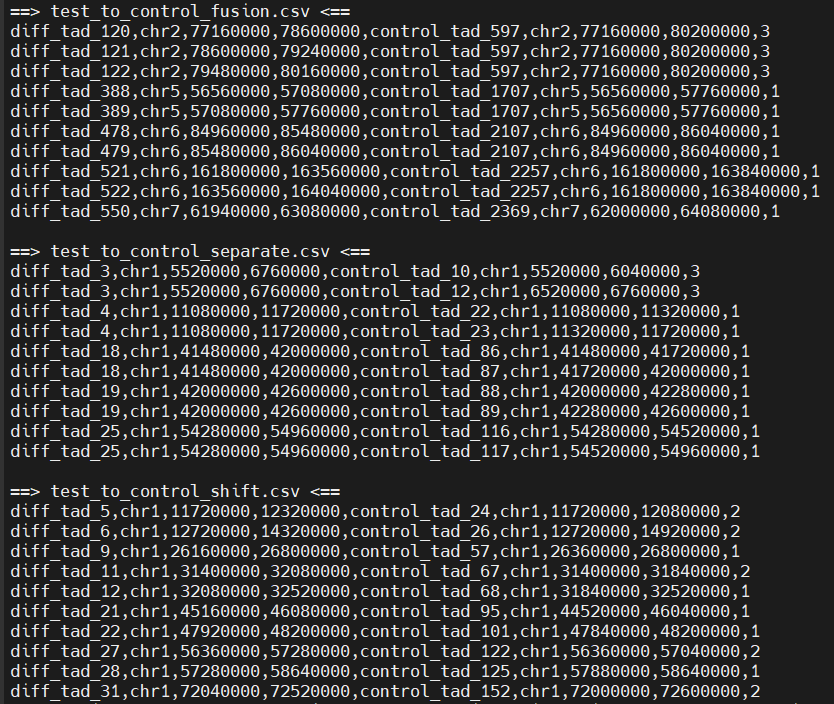
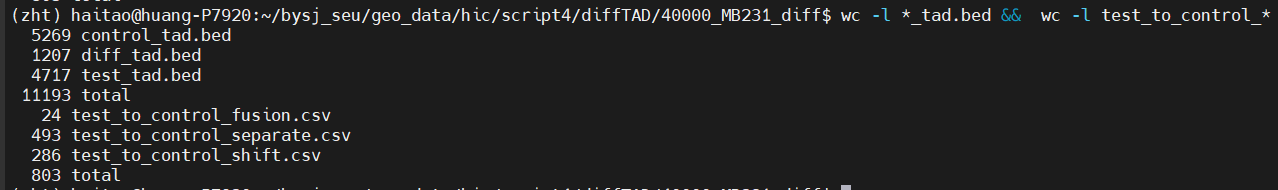
可以发现主要是这3类TAD,还有这3类之外的TAD没有进行处理,不过重点就focus在这3类上了
注意理解掌握算法原理,如何计算以及标注这些对应的边界信息等,不是传统的1维bed边界计算
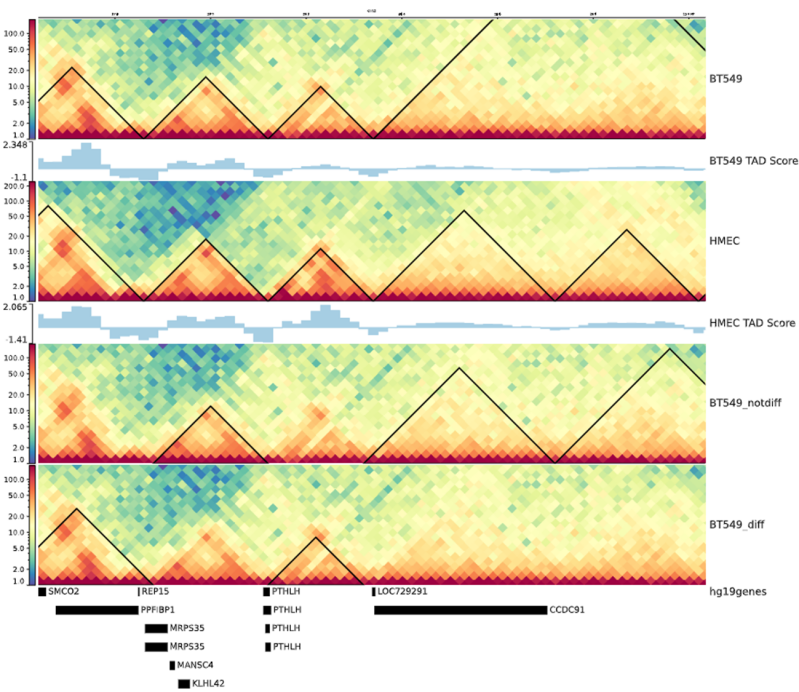
(2)hicexplorer分析差异TAD模块:
继续使用hicexplorer中的函数hicInterIntraTAD:
https://hicexplorer.readthedocs.io/en/latest/content/tools/hicInterIntraTAD.html
提取和计算不同的外部间和内部TAD值和比率,仅仅作为出图展示
类似于neighbor分析,需要解读分析:
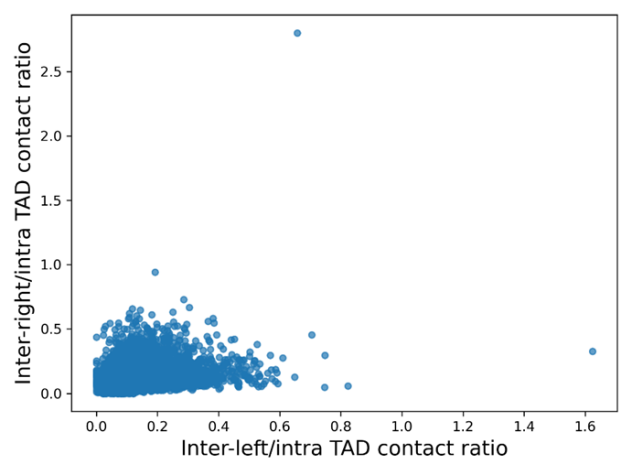

参考HiC_pipeline/pipeline/hic 基础pipeline骨架/06_hicInterIntraTAD.sh
再看看hicexplorer中还有没有什么能够分析TAD的工具
(3)TAD绘图展示:主要使用hicexplorer中工具hicPlotTADs
也可以试试看之前的工具,**看看已经call出来的TAD是否能够配合到前面的juicebox track
使用juicebox或者之前对应的higlass软件以及其他的一些能够直接绘图的hic软件**
下面开始使用hicPlotTADs函数(可以试一下hicplotmatrix)
基本参考:
https://hicexplorer.readthedocs.io/en/latest/content/tools/hicFindTADs.html
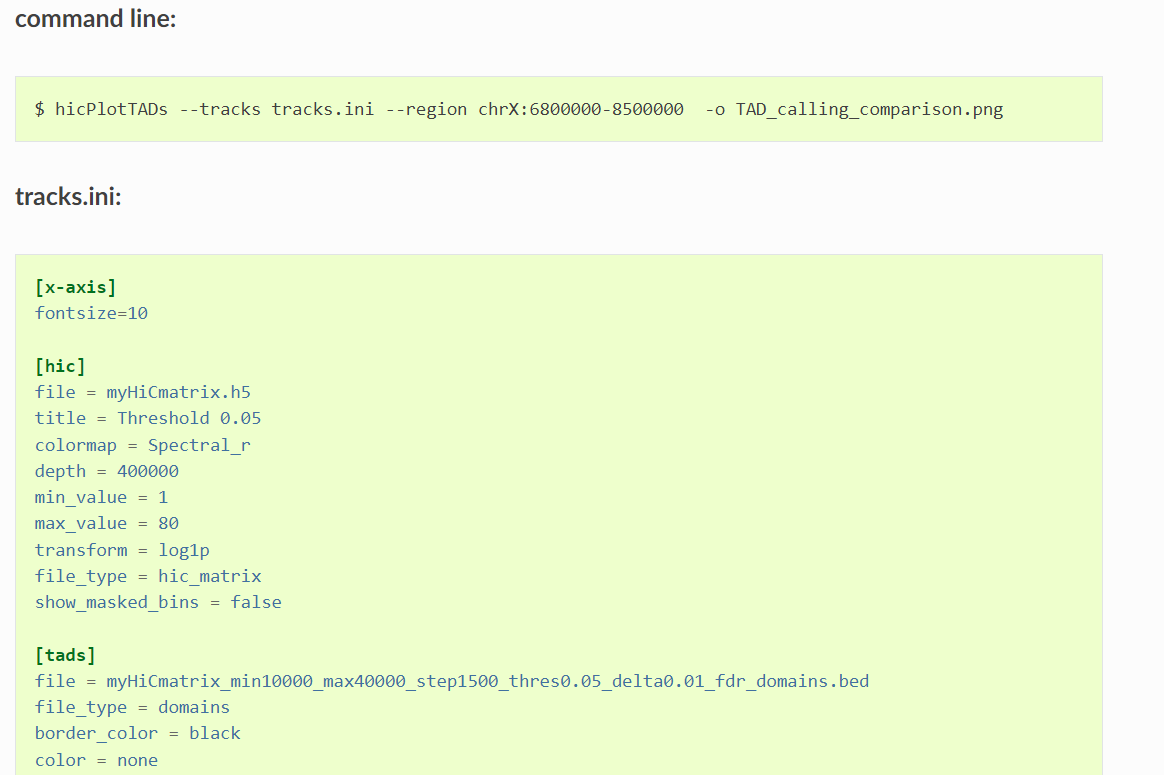
https://github.com/dozmorovlab/UO_project_2021/tree/main/05_differential_tads
注意,绘图的ini文件中最好不要加注释,可以编写一个脚本,将绘图配置文件ini以及各种可选参数作为命令行参数输入,写一个普适性比较强的脚本;
整体脚本参考https://github.com/MaybeBio/TNBC-project/blob/main/3%2CTAD/plot%20TAD/plot_TAD.sh或
https://github.com/MaybeBio/TNBC-project/blob/main/3%2CTAD/plot%20TAD/plot_TAD_2.sh
即HiC_pipeline/pipeline/hic 基础pipeline骨架/05_plot_TAD.sh、pipeline/hic 基础pipeline骨架/05_plot_TAD_2.sh。
绘图的track设置文件ini参考:
https://hicexplorer.readthedocs.io/en/latest/content/tools/hicFindTADs.html
https://github.com/dozmorovlab/PDX-HiC_processingScripts/blob/main/8.TADs-plotsettings.ini
https://github.com/dozmorovlab/UO_project_2021/blob/main/05_differential_tads/tracks.ini
以及参考https://github.com/MaybeBio/TNBC-project/tree/main/3%2CTAD/plot%20TAD中

可以搭配辅助的track:
CTCF等蛋白结合的bigwig文件
loop的bedGraph文件
区室的bedGraph文件
其他track的bed或者是bw文件
感兴趣顺式调控元件EPC的bed或者是bw文件等
绘图这里其实就可以参考后续研究中与转录相关的感兴趣的gene等位点了,
比如说是SE的bed文件,CRC的bed文件等
GWAS发现的风险位点等:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02898-w#Sec29

hg参考gene的ref位点的bed文件:
https://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/

可以直接到ucsc的table浏览器上下载,但是暂时没有找到symbol的,都是ncbi或者是ucsc的自己的gene id,或者是ens
虽然建议下载ensemble,然后上面也可以找到id转换的文件,总之什么ref都可以,
但是效果像下面这样很不好,有空可以自己id转换一下,
下面直接使用dchic中自动生成的hg19的symbol id的bed文件
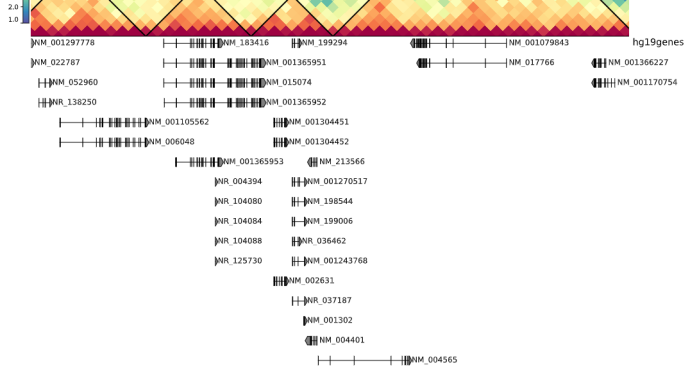
当然这个refgene的bed有issue相关:
https://github.com/ay-lab/dcHiC/issues/73
(4)其他TAD的统计分析等:
HiC_pipeline/pipeline/hic 基础pipeline骨架/07_TAD_Width.sh
其中统计脚本https://github.com/magmarshh/TADWidth?tab=readme-ov-file
待补充
(5)GENOVA分析TAD模块:
主要设置以及文件导入依然参考compartment分析部分的Rmd文件
https://github.com/MaybeBio/seu-TNBC-project/blob/main/2%2CCompartment/ABsaddle.Rmd(1700行以下)
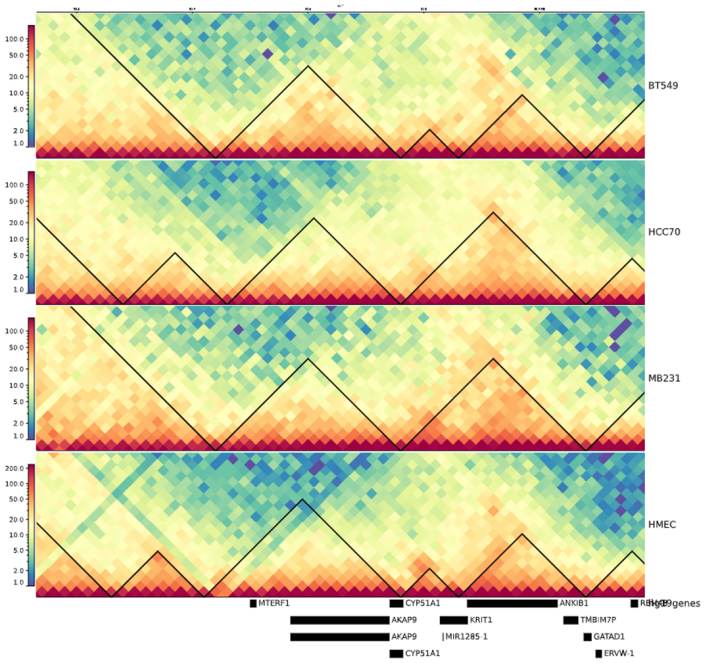
整理后见HiC_pipeline/pipeline/hic 基础pipeline骨架/08_TAD_genova.Rmd
首先是官方建议方面:


①绝缘评分分析:
本质上其实就是TAD识别的一种算法,所以

②边界绝缘信号分析(绝缘评分)+其他组学track:理论上可以搭配IGV以及juicebox:
本项目使用HMEC中的TAD边界bed作为比对基础,分析该bed坐标在TNBC中的情况,主要是看该坐标在TNBC中的绝缘评分,看有没有降低,以及搭配其他CTCF(TAD边界的一种常见保守信号)以及H3K27ac信号(主要是SE等),但是需要注意:用的是什么bed边界坐标,是HMEC的TAD边界还是TNBC的TAD边界(这个很重要,TNBC数据nature文献是以HMEC的bed作为基础对照,实际上分析的可能不是TNBC的边界,而只是HMEC的边界这一块坐标在TNBC中会怎么样变化)!!!
另外需要注意的是hicexplorer以及GENOVA都能够识别TAD,所以都能够提供边界bed文件,理论上来说如果要统一分析的话那识别TAD就应该使用GENOVA全套的内容

使用的TAD边界bed文件有hicexplorer中的,也有genova中自己的产生的;
各自效果不同,建议统一即可:


一些絮絮叨叨
前者是hicexplore中的,后者是genova中的,前者的效果就是在TAD边界上有局部最低的绝缘评分,但是使用genova绘制的图却没有这个效果,而且很奇怪
实际上后者绘制的效果和前者hicexplorer中使用domains.bed文件一样的效果,我觉得应该是GENOVA中自己写代码将domains与boundary的bed文件1混淆了,重新修改再跑
其实查看hicexploer中跑出来的结果:

domain和boudary的bed文件实际上是差不多的,
不对,实际上看一下,其实边界bed文件中比较窄,而且边界差实际上就是res大小,但是domain的bed实际上范围更大,所以所以实际上两者不能够混淆,
该使用边界bed文件的地方就应该使用hicexplorer中的边界的bed文件
该使用domain的bed文件的时候就应该使用genova中自身的TAD call或者是hicexplorer中的domaind的bed文件!!!!!!!!!!
应该是边界以及TAD domain都是有宽度的,但是两者的宽度不是一个级别,明显domain的宽度即TAD的大小肯定是比边界大的
hicexplorer中获得的文件肯定是行的,无论是边界还是domain文件,
但是都需要转换
但是genova中自身获得的只有TAD的bed文件
我如果给的是genova自身TAD的bed文件+center,那看的确实是domian内部的绝缘评分,而不是TAD边界的评分,
但是如果bed_pos修改为start或者是end的话,
实际上查看hicexplorer中所获得两个bed文件就可以看出,
TAD的起点或者是终点就是边界的中点,边界大小实际上就是TAD起点或者是中点左右衍生一半res数据而已
所以看绝缘评分的时候,给出边界的bed文件,但是给出center;与给出domian的bed文件,但是给出start或者是end的效果是一样的,只不过周围绘制的时候给出domian肯定会比边界更长距离
而且因为domian的边界是相互连接的,即连续的数据
所以我的genova中本身call出来的结果中就是这种效果,就是因为是domain文件,
所以可以使用【1:3】来获取边界信息

或者是bed_pos中选用start或end,都可以绘制边界上的信号
所以
如果要看TAD上domain全局的信号:
可以使用hicexplorer中的domain的bed文件,或者是genova中自身call 出来的TAD就像上面那张图那样
如果要看TAD边界上的信号:
可以使用hicexploer中边界的bed文件,或者是genova中自身TAD call出来的选用【1:3】,或者是bed_pos修改为start或end
我期望的效果是这样的:

至于效果,可以从绝缘评分的bio意义出发,就说是边界的绝缘能力得到了削弱。
另外:
比对bed信号的话,如果没有问题,一律使用HMEC的bed信号,TNBC的bed信号就暂时不使用了;

①主要是TAD边界以及domain内部的bed文件
边界好说,主要是如何进行统计检验p
以及domain内部的绝缘评分如何进行分析?
②寻找其他bed文件:
使用增强子的bed文件,包括allE以及TE以及SE的bed文件
但是最后的绘制效果并没有明显的生物学意义以及推论
首先是使用HMEC中的allE+TE+SE文件进行分析比较,然后其次是TNBC的同理
SE区域是绝缘评分的局部极值点极大值
另外还有CTCF的bed文件,
但是TNBC使用的bed文件是分散的,可以取交集,或者是取并集
对于3个narrowpeak文件:
可以效仿前面对应的SE的文件一样,直接取交集:
此处直接选择合并之后取并集


但是发现则这样的话数据差异会非常大
效果还可以,但是不知道如何解释,只能说与之前研究一致——但是需要说明结果的一致性以及唯一性:
还是需要一个TAD尺寸的小提琴图说明,然后使用区域以及边界的信号说明只有边界的绝缘评分是最低的,
所以后面CTCF评分如果也拉长到全chr上的尺度也能说明最后结果就是边界上富集CTCF
还有一个就是k27ac的信号:

可以在40kb内部再做一个相应的CTCF之类
然后边界之类互作可以使用RCP+定量化
可以将TAD边界以及其他track的bed文件都整一个远超于尺寸得范围,
比如说TAD是连续的,然后整个区域上就边界最低
然后其他的区域可以同样,比如说如果SE全尺度内是最低的,那就说明TAD边界富含SE这个结论



絮絮叨叨完毕
③ATA分析:聚集TAD分析,还是②中的问题,本项目处理的是bulk TAD,事实上在分析出差异TAD之后应该按照差异TAD进行分类,然后对不同类的TAD进行聚集分析之类
通过在TADs周围聚集Hi-C基质,可以在全球范围内研究TADs。在总体TAD分析(ATA)中,由于TAD有不同的大小,所以它们被重新缩放到一个统一的大小,然后将结果在整个基因组中平均

解读分析:总之是将全基因组范围内TAD周围的hic互作矩阵聚集在一起,因为TAD具有不同的大小,所以将其统一缩放到一个统一的大小
显示所有TADs及其周围的平均接触分布;

同样像区室一样,可以对结果进行定量化quantify再分析,
GENOVA中有很多函数是通用的,比如说quantify等需要注意
絮絮叨叨

使用10kb的数据,并进行后续的定量化分析,绘制相应的ggstat的p检验的图
其实一直有一个奇怪的地方,就是识别出来的TAD是连续的,当然TAD在chr不是全部都有
但是识别出来的TAD就是连一块的连续区域


有个问题corner减弱,但是TAD邻居增强
以及比边界文件实际上是连续的
也不对,换个思路,使用的全都是HMEC中的TAD文件
絮絮叨叨结束
④TAD+N分析:即intra内部互作,与inter外部邻居互作比较,本质上还是边界分析
此处的neighbor分析,以及ATA分析和绝缘评分分析,以及上面hicexplorer中的hicInterIntraTAD分析,结合起来,可以分析TAD边界的破坏与否
 TAD+N分析:即intra内部互作,与inter外部邻居互作比较
TAD+N分析:即intra内部互作,与inter外部邻居互作比较
可以结合前面hicexploer中绘制出来的邻居互作比例(hicInterIntraTAD),本质上还是边界分析

絮絮叨叨
边界+neighbor分析

可以绘制这个文件的散点图,xy轴分布+线性回归


絮絮叨叨结束
⑤其他一些统计指标:
比如说是size统计,
3,可以深入分析的模块:
(1)差异TAD部分:
可以参考UO_project_2021/05_differential_tads/
尤其是separate与fusion之类的TAD,可以在对应的边界上分析CTCF的结合情况以及对应motif方向的分析;注意差异TAD有很多类型,不是笼统地在所有TAD边界上分析
(2)进一步结合SE以及CRC部分
(3)TAD对于转录调控的影响:自己本来的计划可以从pre3中看看内容todo
https://github.com/robinweide/GENOVA/issues/272参考,
至于·TAD层面的转录,主要是TAD差异的对象做不了什么特异性的分析,
如果要寻找差异的TAD对象,可能是边界shift,fusion,或者内部de novo子TAD生成,
但是gene还是gene,和边界无关,
和TAD相关,但是TAD不好定量,
要找特异性TAD内部的gene,最后还是要转入到loop层面
边界分析主流是enhancer劫持,那还是loop分析


















 2023
2023

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









