







SCENIC+:增强子和基因调控网络的单细胞多组学推理
大致走马观花的学习了一下这篇文章,其中knockdown simulation这是一个生物和计算结合的名词,可用于模拟某一转录因子的敲低意义,可联合分子生物实验。
摘要
对单个细胞中染色质可及性和基因表达的联合分析为破译增强子驱动的基因调控网络(GRN)提供了机会。在这里,我们提出了一种用于推理增强器驱动的 GRN 的方法,称为 SCENIC+。 SCENIC+ 预测基因组增强子以及候选上游转录因子 (TF),并将这些增强子与候选目标基因联系起来。为了提高 TF 识别的召回率和精确度,我们策划并聚类了一个包含 30,000 多个主题的主题集合。我们在不同物种的不同数据集上对 SCENIC+ 进行了基准测试,包括人外周血单核细胞、ENCODE 细胞系、黑色素瘤细胞状态和果蝇视网膜发育。接下来,我们利用 SCENIC+ 预测来研究大脑皮层中人类和小鼠细胞类型之间的保守 TF、增强子和 GRN。最后,我们使用 SCENIC+ 研究基因调控沿分化轨迹的动态以及 TF 扰动对细胞状态的影响。 SCENIC+ 可在 sceneplus.readthedocs.io 上获取。
Introduction
细胞身份由基因调控网络(GRN)编码,其中转录因子(TF)与顺式调控元件(CRE)组相互作用以控制靶基因的转录。 CRE 通常具有细胞类型特异性,并由特定的 TF 结合位点 (TFBS) 组合组成。深入了解 GRN 对于理解发育、进化 、和疾病的生物学方面非常重要;然而,对顺式监管层面的 TF 与目标关系的了解仍然有限。
包括染色质免疫沉淀和测序 (ChIP-seq) 在内的实验技术已经产生了大量的 TF 结合数据集。然而,对于细胞类型多样性较高的组织,由于需要大量同质细胞,绘制 TFBS 仍然具有挑战性。此外,对于大多数TF来说,缺乏高质量的抗体。最近已经描述了提高细胞分辨率的替代方法(例如,单细胞 CUT&Tag、nano-CT和 NTT-seq)或依赖于基因标记(例如,DamID和 nanoDam),但此类方法仍然难以实现扩展到所有 TF。
计算模型是识别 TFBS 的替代方法。例如,SCENIC 将单细胞 RNA 测序 (scRNA-seq) 共表达网络与 TF 基序发现相结合 11,12,但它无法识别 TF 靶向的确切 CRE,并且仅使用基因假定调控空间的一小部分 13, 14.利用单细胞染色质可及性数据,可以大幅提高 TFBS 预测的准确性15。事实上,细胞类型中特定的基因组区域通常代表增强子,并且富含 TFBS 组合。
在这里,我们开发了 SCENIC+,这是一种计算框架,它将单细胞染色质可及性和基因表达数据与基序发现相结合,以推断增强子驱动的 GRN (eGRN)。enhancer-driven GRNs (eGRNs).
SCENIC+ 使用超过 30,000 个 TF 基序来预测 eGRN
SCENIC+ 是一个三步工作流程,包括识别候选增强子、识别富集的 TF 结合基序以及将 TF 连接到候选增强子和目标基因(Fig. 1a)。输出是一组增强子驱动的调节子 (eRegulons),形成 eGRN。
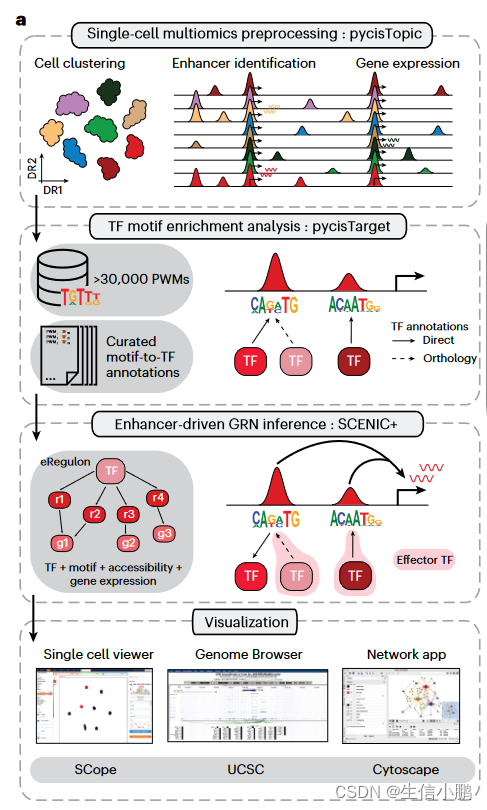 Fig. 1 | The SCENIC+ workflow and motif collection. a, SCENIC+ workflow. Topics and DARs inferred with pycisTopic are transformed into cistromes of directly bound regions by identifying modules that present significant enrichment of the regulator’s binding motif using pycisTarget. SCENIC+ integrates region accessibility, TF and target gene expression and cistromes to infer eGRNs, in which TFs are linked to their target regions and these to their target genes. PWM, position weight matrix; UCSC, University of California, Santa Cruz.
Fig. 1 | The SCENIC+ workflow and motif collection. a, SCENIC+ workflow. Topics and DARs inferred with pycisTopic are transformed into cistromes of directly bound regions by identifying modules that present significant enrichment of the regulator’s binding motif using pycisTarget. SCENIC+ integrates region accessibility, TF and target gene expression and cistromes to infer eGRNs, in which TFs are linked to their target regions and these to their target genes. PWM, position weight matrix; UCSC, University of California, Santa Cruz.
图1| SCENIC+ 工作流程和主题集合。 a、SCENIC+工作流程。通过使用 pycisTarget 识别显着富集调节器结合基序的模块,将使用 pycisTopic 推断的主题和 DAR 转化为直接结合区域的顺反子。 SCENIC+ 整合了区域可访问性、TF 和目标基因表达以及顺反子来推断 eGRN,其中 TF 与其目标区域相连,而这些区域与其目标基因相连。 PWM,位置权重矩阵; UCSC,加州大学圣克鲁斯分校。
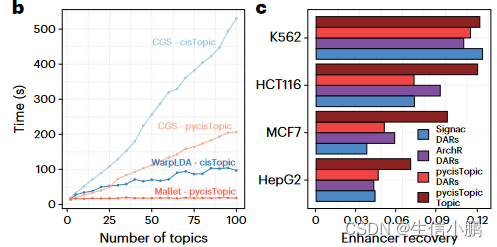
为了找到候选增强子,使用 pycisTopic 对可访问染色质 (scATAC-seq) 数据进行单细胞分析,pycisTopic 是 cisTopic 的更快的 Python 重新实现(图 1b 和扩展数据图 1a-f)。 SCENIC+ 使用差异可访问区域 (DAR) 和主题、跨细胞类型或状态的共同可访问区域集作为增强子候选。与 DAR 相比,功能增强子区域的主题更加丰富(图 1c 和扩展数据图 1g)。
为了发现候选增强子中潜在的 TFBS,我们利用基序富集分析。为此,我们创建了迄今为止最大的主题集合(补充说明 2),并构建了一个名为 pycisTarget 的 Python 包。 pycisTarget 实现了两种用于主题富集分析的算法:基于 cisTarget 排名和恢复的算法,和称为主题差异富集 (DEM) 的 Wilcoxon 秩和测试(补充说明 3)。
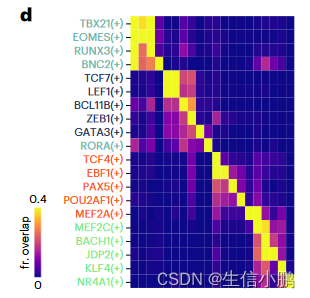
motif collection是一个二级数据库,包含从 29 个集合收集的 32,765 个独特motif (图 1d 和扩展数据图 2a、b)以及 TF 注释。该集合在人类、小鼠和果蝇中分别包含总共 1,553 个 TF、1,357 个 TF 和 467 个 TF(图 1e 和扩展数据图 2c)。我们根据基序与基序的相似性对所有基序进行聚类,发现与每个簇使用单个“原型”基序相比,使用簇内的所有基序对候选区域进行评分可产生显着更高的精度和召回率(图 1f 和扩展数据图) .2d–h)。 cisTarget 和 DEM 算法都优于 Homer(图 1f 和扩展数据图 2d),DEM 算法能够检测具有相似主题内容的区域集中的差异主题(扩展数据图 2i-j)。
SCENIC+ 接下来使用 GRNBoost2(参考文献 23)来量化 TF 和候选增强子对目标基因的重要性,并使用线性相关性推断调控方向(激活/抑制)。使用第二次富集分析将基序富集分析结果与 GRNBoost2 推论相结合,以恢复每组基序的最佳 TF。这就形成了 eRegulon,一种具有一组目标区域和基因的转录因子。 a TF with its set of target regions and genes.
对于最小和最大的测试数据集,工作流程的总体运行时间和内存范围分别为 1 小时和 21 Gb 到 44 小时和 461 Gb(扩展数据图 3)。
SCENIC+ 在 PBMC 多组数据上的图示
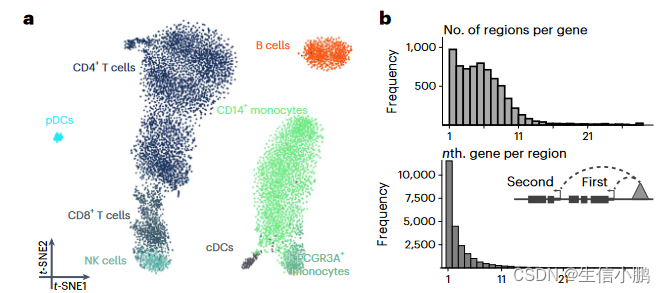
我们首先分析了包含 9,409 个人外周血单核细胞 (PBMC) 的公开单细胞多组学数据集,以展示和验证 SCENIC+。基于 eRegulon 富集分数的降维分离了主要的生物细胞状态(图 2a)。 SCENIC+ 鉴定了 53 个激活剂 eRegulons,总共针对 23,470 个区域和 6,142 个基因。总共 89% 的基因具有 1-10 个预测增强子,并且 49% 的增强子预计最有可能调节最近的基因(图 2b)。
SCENIC+ 包括了著名的 B 细胞(EBF1、PAX5 和 POU2F2/POU2AF1)、T 细胞(TCF7、GATA3 和 BCL11B)、自然杀伤 (NK) 细胞(EOMES、RUNX3 和 TBX21)、树突状细胞(SPIB 和 IRF8)的主调节因子)和单核细胞(SPI1 和 CEBPA)(图 2c)。
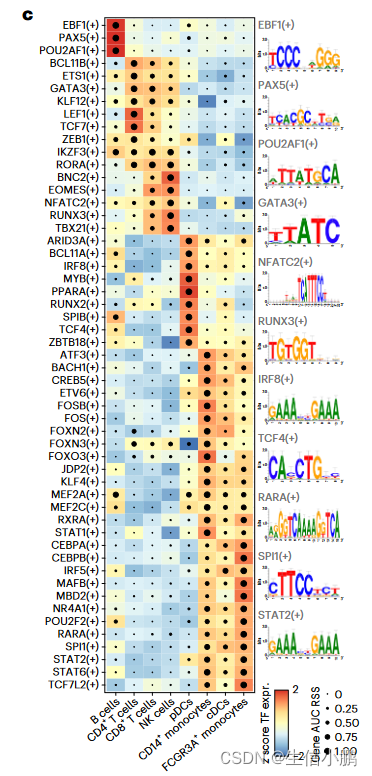
前 5 个细胞类型特异性 TF 中的大多数都显示出与共享增强子的共结合。对于对同一细胞类型不具有特异性的 TF,没有观察到这种协同性(图 2d)。
特别是,对于 B 细胞,SCENIC+ 表明 EBF1(+)、PAX5(+) 和 POU2F2/AF1(+) 之间存在协同作用(图 2e),其大多数预测的目标增强子与 EBF1、PAX5 和 POU2F2 ChIP 有很强的Chip-seq数据(图2f-g)。
总之,SCENIC+以高通量的方式推断出不同PBMC类型的关键调控因子以及这些调控因子的基因组目标区域。可以利用这一点来推断协作性。
SCENIC+ prioritizes functional enhancers (SCENIC+ 优先考虑功能增强剂)
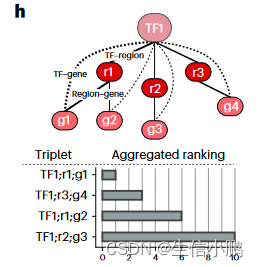
SCENIC+ 对 TF-基因、区域-基因和区域-基序评分使用自动阈值处理程序,以获得离散的 eRegulons 集合;然而,在某些情况下,根据 TF、区域和基因的重要性对其进行排名可能会有所帮助。为此,我们实施了对 TFregion-gene 三联体进行定量排名的排名。该排名是 TF-区域得分、TF-基因得分和区域-基因得分的综合排名(图 3h)
我们测试了三元组排名是否可用于优先考虑潜在的增强剂。事实上,前 10% 的三联体区域具有更高的 ChIP-seq 信号(通过 Enformer 进行实验和计算机模拟测量)以及更高的增强子活性(通过 STARR-seq 测量)(图 3i)。
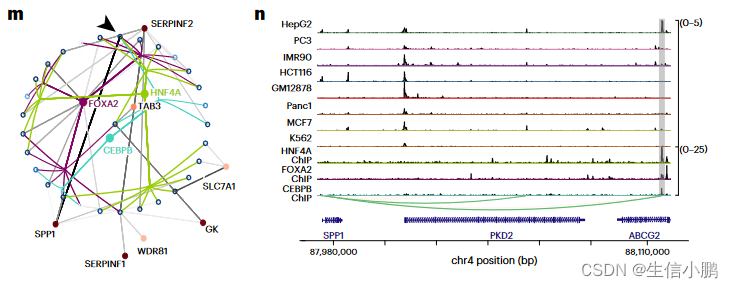
为了进一步说明这一点,我们重点关注 HepG2 细胞的三个主要调节因子:HNF4A、FOXA2 和 CEBPB。这些 TF 的预测目标区域对于这些 TF 具有较高的 ChIP-seq 覆盖率,正如 Enformer 通过实验和计算机测量所测量的那样(图 3j-l)。与覆盖率低的区域相比,ChIP-seq 覆盖率高的区域也具有更高的 TF-to-region 排名(图 3j-l)。相比之下,GRaNIE、Pando 或 CellOracle 对相同 TF 的预测目标区域非常稀疏。仅对于 HNF4A,GRaNIE 预测的目标区域与 SCENIC+ 的预测目标区域吻合良好;然而,GRaNIE 识别出 Enformer 得分较低的目标区域子集,尽管它们与 ChIP-seq 峰重叠(图 3j)。
接下来,我们放大了根据三联体排名预测最重要的 HNF4A 目标区域。该区域也预计会成为 FOXA2 和 CEBPB 的靶点,并预计会调节 HepG2 细胞的标记基因 SPP1(图 3m)(平均对数倍数变化 (FC) 为 9.24)。该区域在 HepG2 细胞中特别容易进入,并且对 HNF4A、FOXA2 和 CEBPB 具有高 ChIP-seq 信号(图 3n)。总而言之,该区域是 HepG2 中 SPP1 的强大增强子候选区域。
SCENIC+ simulates phenotype switching of cancer cell states (SCENIC+ 模拟癌细胞状态的表型转换)
阅读后,自我感觉,这一部分是很重要的一个应用。提出knockdown simulation这个概念,可以和分子生物实验向结合。
癌细胞的基因调控网络分析有望识别稳定(吸引子)细胞状态及其调节因子。作为案例研究,我们对代表不同黑色素瘤状态的九种黑色素瘤细胞系进行了 scATAC-seq,并将这些数据与之前发布的相同细胞系的 scRNA-seq 数据相结合。
根据 eRegulon 富集分数,细胞聚集在三种状态(图 4a)。此外,基于 SCENIC+ 网络中前 25% TF 到 TF 边缘的布尔模型 49 足以概括所有主要单元状态(扩展数据图 8a、b)
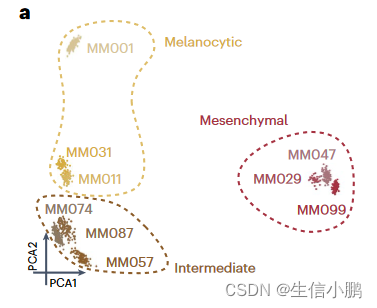
Fig. 4 | SCENIC+ analysis using separate scATAC-seq and scRNA-seq data on a mix of human melanoma lines. a, PCA of 936 pseudo-multiome cells based on target gene and target region enrichment scores.
图4|使用单独的 scATAC-seq 和 scRNA-seq 数据对人类黑色素瘤系的混合物进行 SCENIC+ 分析。 a,基于目标基因和目标区域富集分数的 936 个伪多组细胞的 PCA
SCENIC+ 恢复了已知的黑素细胞 (MEL) 状态(MITF、SOX10、TFAP2A 和 RUNX3)、间充质 (MES) 状态(JUN、NFIB 和 ZEB1)以及由 MEL TF 控制的 MEL 中间亚状态的调节因子,并辅以SOX6、EGR3 和 ETV4(图 4b 和扩展数据图 8c-g)48,50,51。之前有人提出 RUNX 基序是 MEL 增强子代码 的一部分,但 RUNX 家族的哪个成员尚不清楚。使用 SCENIC+,我们预测它很可能是 RUNX3(图 4b)。
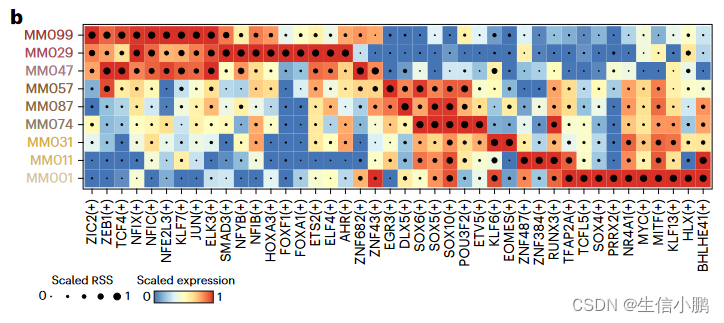 b, Heat map/dot-plot showing TF expression of the eRegulon on a color scale and cell-type specificity (RSS) of the eRegulon on a size scale. b,热图/点图显示了 eRegulon 在颜色尺度上的 TF 表达和 eRegulon 在尺寸尺度上的细胞类型特异性 (RSS)
b, Heat map/dot-plot showing TF expression of the eRegulon on a color scale and cell-type specificity (RSS) of the eRegulon on a size scale. b,热图/点图显示了 eRegulon 在颜色尺度上的 TF 表达和 eRegulon 在尺寸尺度上的细胞类型特异性 (RSS)
众所周知,黑色素瘤细胞可以动态地将状态从 MEL 转变为 MES,反之亦然,从而驱动转移和治疗耐药,这一过程称为表型转换 52。特定转录因子的敲除可以推动这一过程48。
为了模拟表型转换并优先考虑此过程中的 TF,我们从 CellOracle和 GRaNPA中汲取灵感,使用 SCENIC+ 作为特征选择方法,并为每个基因训练随机森林 (RF) 回归模型,以根据表达来预测其表达他们的上游 TF。拟合模型后,我们通过将 TF 的表达式设置为零来使用它来预测 TF 扰动的效果。为了考虑间接影响(针对其他 TF 的 TF),扰动的基因表达值会经过多次迭代传播。模拟扰动的效果可以通过将模拟基因表达矩阵与原始基因表达矩阵共同嵌入来可视化(图4c)。
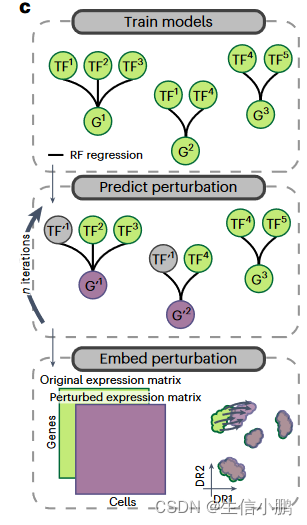
c, Illustration of how predictions from SCENIC+ can be used to simulate TF perturbations. Top: SCENIC+ is used as a feature selection method and RF regression models are fitted for each gene using TF expressions as predictors for gene expression. Middle: the expression of TF(s) is altered in silico and the effect on gene expression is predicted using the regression models, which is repeated for several iterations to simulate indirect effects. Bottom: the original and simulated gene expression matrices are co-embedded in the same dimensionality reduction to visualize the predicted effect of the perturbation on cell states.
c,说明如何使用 SCENIC+ 的预测来模拟 TF 扰动。上图:SCENIC+ 用作特征选择方法,并使用 TF 表达作为基因表达的预测因子来拟合每个基因的 RF 回归模型。中:在计算机中改变 TF 的表达,并使用回归模型预测对基因表达的影响,该模型重复多次迭代以模拟间接影响。底部:原始基因表达矩阵和模拟基因表达矩阵共同嵌入相同的降维中,以可视化扰动对细胞状态的预测影响。
这里提及到的这个理念还是很有趣的,通过原始数据,通过随机森林的方法为TF和基因之间构建一个模型,预测基因的上游转录因子。然后,人为将TF转录因子表达设置为0,查看基因表达情况变化。
这个流程的真正意义在于,通过这样的方法,在没有进行试验的情况下,我们就可以进行基因的敲低的检验和探索。这应该是一项很有意义的工作。knockdown simulation这是一个生物和计算结合的名词
作为原理证明,我们模拟了 SOX10 KD 对 MEL 状态的影响。值得注意的是,SOX10 KD 后的模拟细胞表明它们切换到更像 MES 的状态,即 MES 基因上调,MEL 基因下调,并且这种效应在四次模拟迭代后稳定下来(图 4d)。 SOX10 敲低的这种预测效应在中间细胞系中最强,并且由实验 SOX10 KD 完全重现,其次是 RNA-seq (图 4e 和扩展数据图 8d)。
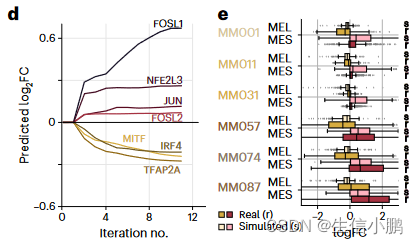
d, Predicted logFC of mesenchymal (red shades) and melanocytic (yellow shades) marker genes over several iterations of SOX10 knockdown simulation.
d,在 SOX10 敲低模拟的多次迭代中预测的间充质(红色阴影)和黑素细胞(黄色阴影)标记基因的 logFC。
受到这一结果的鼓舞,我们模拟了所有已识别的转录因子的扰动。 RUNX3、SOX10 和 MITF 的模拟敲低显示出将细胞从 MEL 切换到 MES 的最强潜力;而ZEB1、SOX4或SMAD3的敲低预计会导致从MES到MEL的反向转换(图4f-g),这与这些TF在上皮间质转化(EMT)中的作用一致。我们还确定了MXI1 和 ZNF487 作为潜在的 EMT 调节剂,值得进一步研究。因此,该策略可用于优先考虑调节细胞状态和状态转换的 TF。
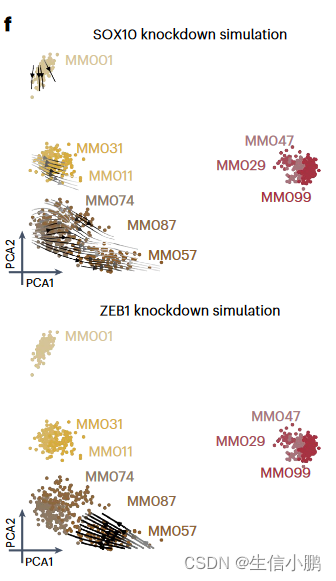
f, Simulated shift after SOX10 and ZEB1 knockdown represented using arrows. Arrows are shaded based on the distance traveled by each cell after knockdown simulation.
f,SOX10 和 ZEB1 敲低后的模拟转变用箭头表示。箭头根据模拟敲低后每个细胞行进的距离进行着色。
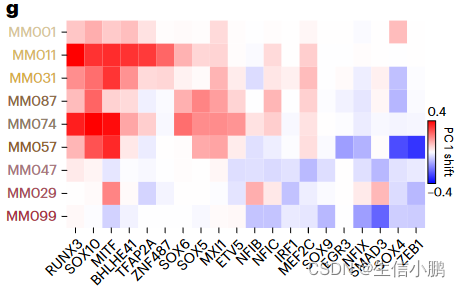
g, Heat map representing the shift along the first principal component of each melanoma line after simulated knockdown of several TFs.
g,热图,表示在****几个 TF 后沿着每个黑色素瘤线的第一主成分的移动。
哺乳动物大脑中 eGRN 的保守性和分化
使用 GRN 速度预测驱动分化的 TF
单细胞组学数据通常用于在分化等动态生物过程中对细胞进行采样。 RNA速率和MultiVelo等模型试图从这些数据中重建最可能的轨迹;然而,这些方法不包括用于动态建模的基因调控信息。
我们推断 SCENIC+ 得出的调控关系可以提供额外的内在线索来预测细胞状态动态。例如,TF的表达可能先于其结合位点的可及性(accessibility),而染色质的可及性又可能先于靶基因的表达(图6a)。因此,我们开发了一种程序来量化 TF 的假定分化力。在这种方法中,细胞沿着伪时间轴排序,并且每个细胞根据其当前的 TF 表达值以及具有最佳匹配的未来目标基因表达的细胞与其未来细胞相匹配。每个细胞中 TF 的分化力被定义为沿伪时间轴到其未来细胞的距离。这些力可以在任何细胞嵌入的网格上绘制为箭头(图 6a)。
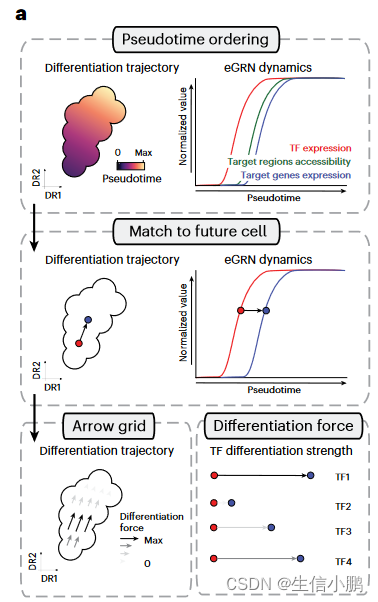 Fig. 6 | Identification of differentiation drivers from SCENIC+ eGRNs. a, Computational approach to infer differentiation drivers from a SCENIC+ analysis. First, differentiating cells are ordered by pseudotime. Second, for each eRegulon, a standardized GAM is fitted along the pseudotime axis for its expression and its target genes (or regions) enrichment scores and each cell in a certain quantile of the GAM TF expression curve is mapped to its future cells in the same quantile in the GAM regulon enrichment curve. Finally, the differentiation force of a cell and regulon is defined as the distance from the TF expression curve to its future cell in the regulon enrichment curve.
Fig. 6 | Identification of differentiation drivers from SCENIC+ eGRNs. a, Computational approach to infer differentiation drivers from a SCENIC+ analysis. First, differentiating cells are ordered by pseudotime. Second, for each eRegulon, a standardized GAM is fitted along the pseudotime axis for its expression and its target genes (or regions) enrichment scores and each cell in a certain quantile of the GAM TF expression curve is mapped to its future cells in the same quantile in the GAM regulon enrichment curve. Finally, the differentiation force of a cell and regulon is defined as the distance from the TF expression curve to its future cell in the regulon enrichment curve.
图 6 |从 SCENIC+ eGRN 中识别差异化驱动因素。 a,从 SCENIC+ 分析推断差异化驱动因素的计算方法。首先,区分细胞按伪时间排序。其次,对于每个 eRegulon,标准化的 GAM 沿着伪时间轴拟合其表达及其目标基因(或区域)富集分数,并且 GAM TF 表达曲线的特定分位数中的每个细胞被映射到相同的未来细胞。 GAM 调节子富集曲线中的分位数。最后,细胞和调节子的分化力定义为调节子富集曲线中从TF表达曲线到其未来细胞的距离。
我们首先将这种方法应用于小鼠大脑中从 少突胶质祖细胞(oligodendrocyte progenitor cells, OPC) 到成熟少突胶质细胞的线性分化轨迹。这揭示了一组保持 OPC 身份的 TF(Olig2、Bcl6 和 Prrx1)。另一方面,Tcf7l2 和 Sox10 在 TF 和靶基因表达之间存在延迟。这可以看作是指向新形成的少突胶质细胞(NFOL)的箭头。最终一组 TF(Zeb2、Meis1 和 Tcf12)被确定为 NFOL 成熟为少突胶质细胞的潜在驱动因素(图 6b)。值得注意的是,Meis1 先前被描述为参与早期神经发生和造血68,但不参与少突胶质细胞的成熟。与此一致,SCENIC+ 还鉴定出 Meis1 是人和小鼠少突胶质细胞中的保守转录因子(图 5c)。
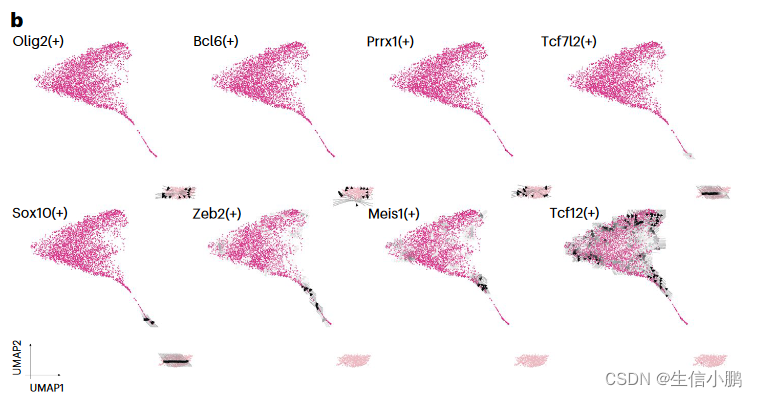
b,沿小鼠皮质中 OPC 分化为成熟少突胶质细胞的箭头网格表示(4,435 个细胞)。
接下来,我们将 GRN 速度应用于发育中的果蝇视网膜中从祖细胞到光感受器或膜间细胞的分支分化轨迹。为此,我们对眼部区域进行了单细胞 (sc) ATAC-seq,并将这些数据与发育中眼睛的 scRNA-seq 和 scATAC-seq 数据相结合(图 6c)14。 SCENIC+ 鉴定出 105 个在眼睛部分活跃的 eRegulons。值得注意的是,SCENIC+ 发现了触角中表达的 Cut (Ct) 抑制因子 eRegulon。已经表明,它是眼域的抑制因子69,在这里我们预测它会直接抑制其他 13 个转录因子,包括 Spineless (ss)、Eyeless (ey)、Twin of eyeless (toy) 和 Optix。
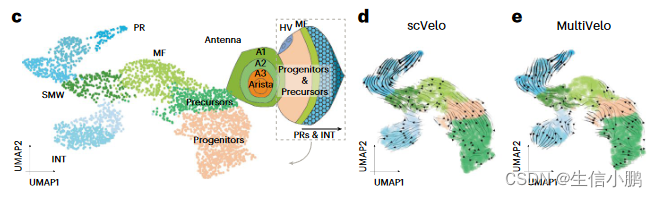
正如 scVelo 和 MultiVelo 推断的那样,细胞遵循从祖细胞到形态发生沟 (MF) 和第二有丝分裂波 (SMW) 的分化轨迹,形成感光细胞 (PR) 或间膜细胞 (INT) 的分支点。图6d、e)。 GRN 速度显示祖细胞中 Optix、Toy 和 Ey 的强分化箭头,其次是 MF 中的 Hairy (hry)、Anterior open (aop)、Rotund (rn) 和 Atonal (ato)。 BarH1 (B-H1)、BarH2 (B-H2)、Sine oculus (so) 和 Glass (gl) 被发现可触发从 MF 向 PR 和 INT 的分化。 Lozenge (lz) 被发现是 INT 身份的关键驱动因素,Tramtrack (ttk) 是其成熟的关键驱动因素。在光感受器分支中,Senseless (sens) 和 Rough (ro) 被确定为分化的关键调节因子。接下来是成熟光感受器中的 Asense (ase)、Lola、Seven up (svp) 和 Scratch (scrt),然后是 Shaven (sv) 和 Onecut (onecut)(图 6f、g)。值得注意的是,这些发现与先前描述的眼盘分化级联一致。
这一部分是GRN结合RNA速率对于发育轨迹推断,并在此基础上,确定对于细胞状态变化过程有关的转录因子。
方法
方法部分很多,作者写的也很详细,
SCENIC+ 工作流程 SCENIC+ 工作流程由三个主要分析步骤组成:(1)从 scATAC-seq 数据中无监督地识别具有共享可访问性模式的增强子; (2)通过基序富集分析预测TFBS; (3) 结合 TF 表达、TFBS、区域可及性和基因表达来预测 eGRN。这些步骤是使用三个 Python 模块执行的:pycisTopic、pycisTarget 和 SCENIC+。详细说明参见补充说明 1。工具、SCENIC+ 代码和教程的链接可在 sceneplus 上找到。 readthedocs.io。
学习文献
Bravo González-Blas, C., De Winter, S., Hulselmans, G. et al. SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks. Nat Methods 20, 1355–1367 (2023). https://doi.org/10.1038/s41592-023-01938-4

















 3617
3617











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











