






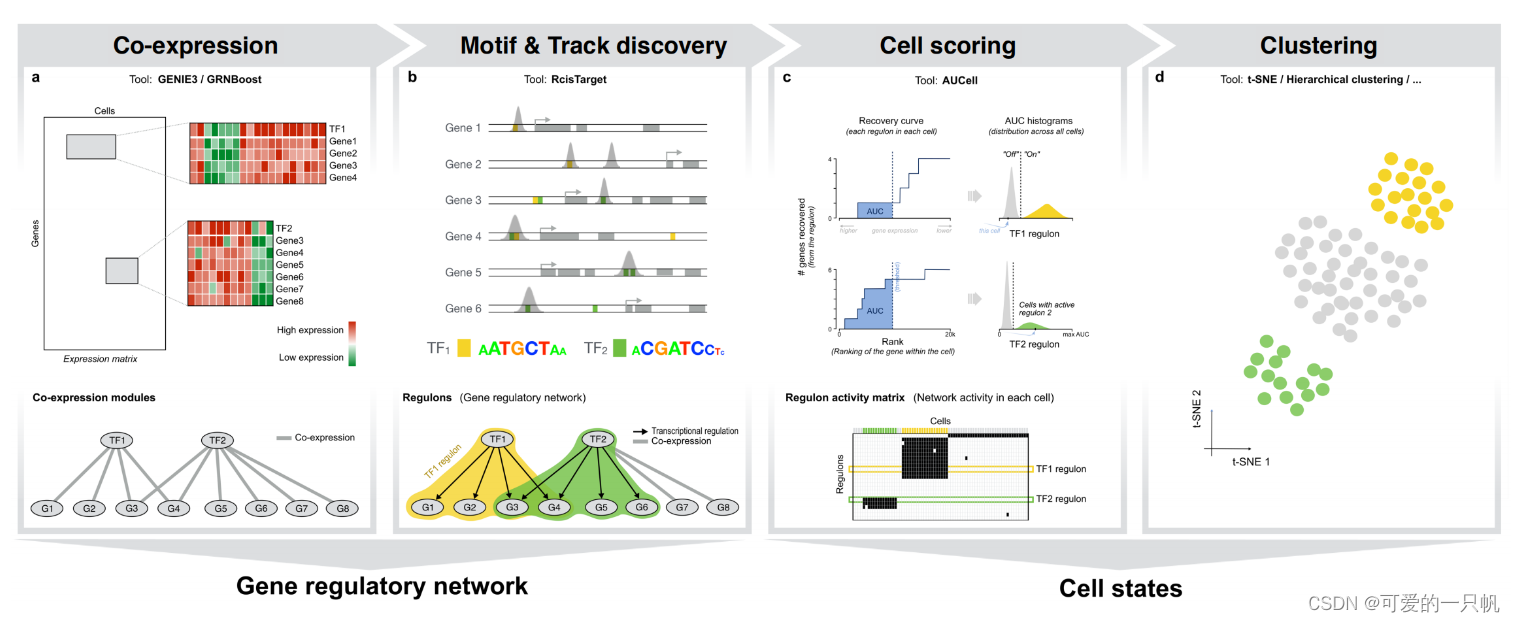
SCENIC (Single-Cell rRegulatory Network Inference and Clustering)是一种能够从单细胞 RNA-seq 数据(SCENIC)或单细胞 RNA-seq+单细胞 ATAC-seq 的组合(SCENIC+, 今年新发的Nature Methods, 2023, 20, 1355–1367)中同时进行转录因子推断、基因调控网络重建的方法。
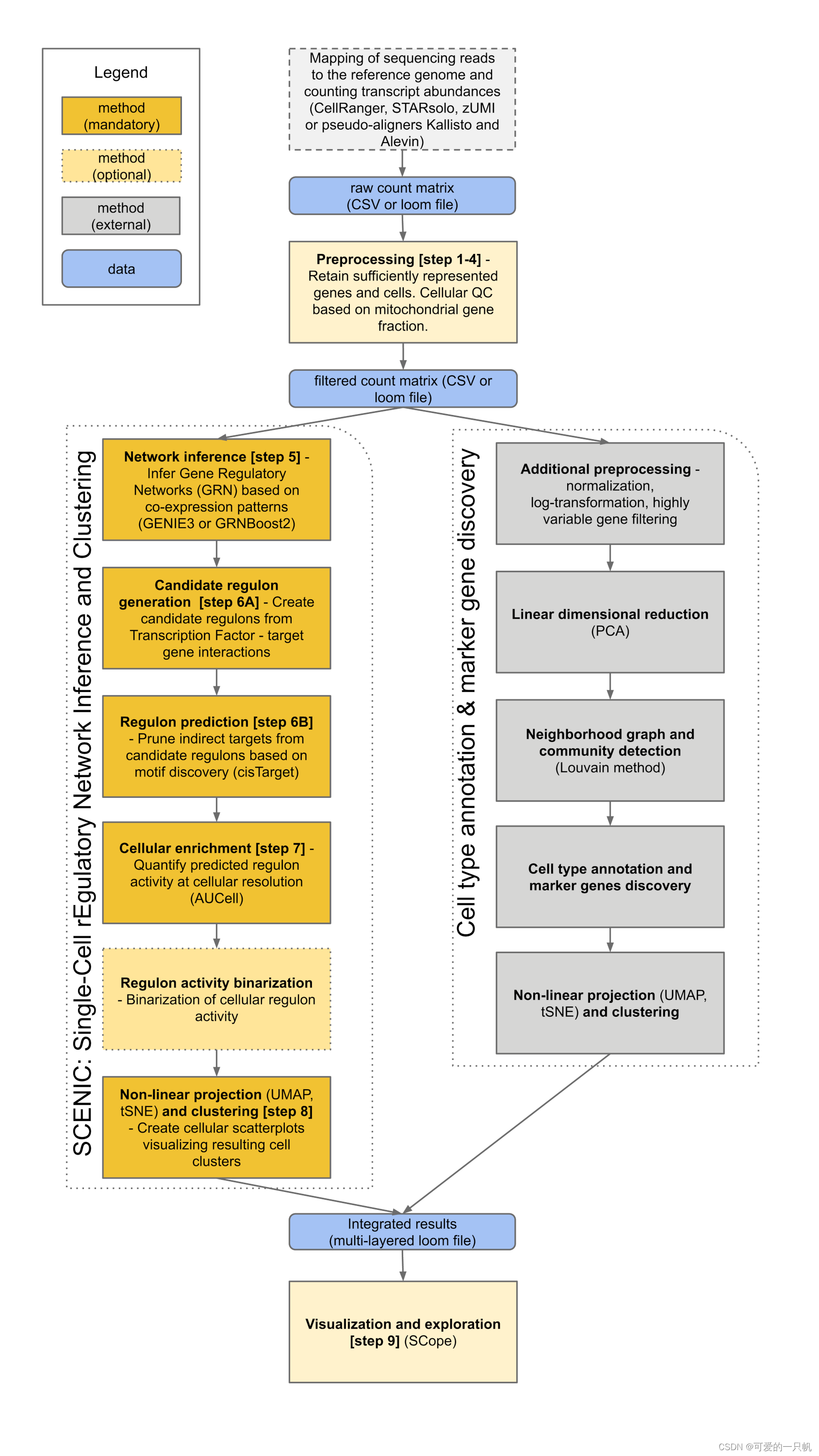
工作流程主要分为三个步骤:共表达网络推断、通过基因 motif 验证的调控网络模块(regulons)、regulons 活性计算。
pySCENIC中共表达推断方法GRNBoost2相比于R中GENIE3有很大的速度提升,所以建议前面的分析使用pySCENIC,后面可视化还是在R中进行(因为本人不怎么会python……)。
1. R中提取转置后的表达矩阵
mat <- as.matrix(sc@assays$RNA@counts)
#mat <- mat[rowSums(mat)>10,]
write.csv(t(mat),file="./counts.csv")
2.conda安装
conda create -n pyscenic python=3.7 #注意自己的python版本
conda activate pyscenic
conda install -y numpy
conda install -y -c anaconda cytoolz
conda install -y scanpy
pip install pyscenic3.制作一个trans.py,主要目的是把提取的表达矩阵转成loom文件,python trans.py运行
import os, sys
os.getcwd()
os.listdir(os.getcwd())
import loompy as lp;
import numpy as np;
import scanpy as sc;
x=sc.read_csv("counts.csv");
row_attrs={"Gene":np.array(x.var_names),};
col_attrs={"CellID":np.array(x.obs_names)};
%lp.create("sc.loom",x.X.transpose(),row_attrs,col_attrs);
4.准备好三个文件ranking database, motif database, TF list(注意物种),下载地址:
Welcome to the cisTarget resources website! (aertslab.org)
pySCENIC/resources at master · aertslab/pySCENIC (github.com)
5.三步搞定,最后得到sc_SCENIC.loom, reg.csv, adj.sc.tsv这三个文件就可以进行下一步可视化了
#推断转录因子与候选靶基因之间的共表达模块
pyscenic grn \
--num_workers 20 \
--output adj.sc.tsv \
--method grnboost2 \
sc.loom \
mm_mgi_tfs.txt
#DNA-motif分析选择TF潜在直接结合的靶点(regulon)
pyscenic ctx \
adj.sample.tsv /mm9-tss-centered-10kb-7species.mc9nr.genes_vs_motifs.rankings.feather \
--annotations_fname /mm10/cisTarget_database/motifs-v9-nr.mgi-m0.001-o0.0.tbl \
--expression_mtx_fname sc.loom \
--mode "dask_multiprocessing" \
--output reg.csv \
--num_workers 20 \
--mask_dropouts
#计算Regulons的活性
pyscenic aucell \
sc.loom \
reg.csv \
--output sc_SCENIC.loom \
--num_workers 8
















 6958
6958

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









