






【Title】Complex Analysis of Single-Cell RNA Sequencing Data
【Publication Book】Biochemistry (Mosc).
【Publication Time】2023 Mar
1.There are several platforms available for scRNA-seq
1.1 Fluidigm C1/Smart-seq
1.2 BD Rhapsody (BD Biosciences, USA)
1.3 Chromium (10x Genomics, USA)
2.The workflow for scRNA-seq
2.1 The cell suspension is prepared by sample homogenization.
2.2 The cells are separated either by physical methods, such as cell sorting or micromanipulation, or via barcoding using plates with oligonucleotides or microfluidics and combinatorics
2.3 The obtained cells are used for the preparation of libraries and following sequencing
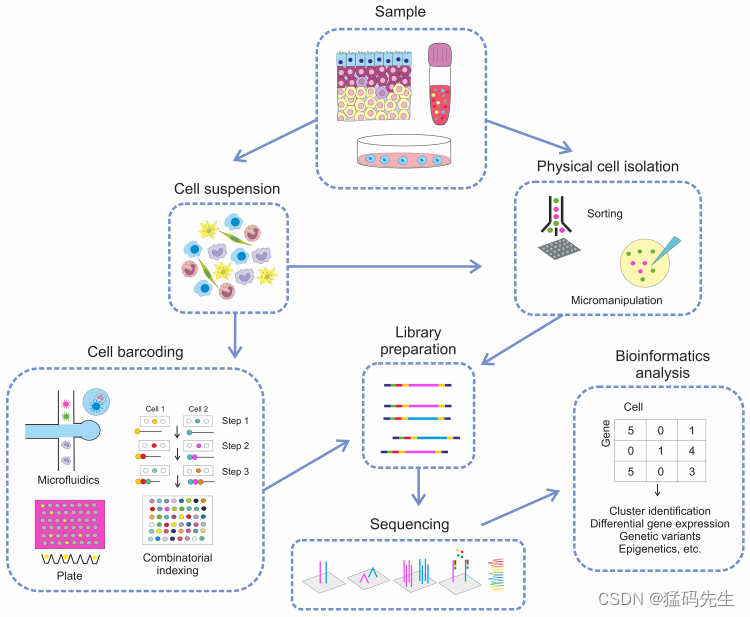
3.The data are processed using bioinformatics techniques
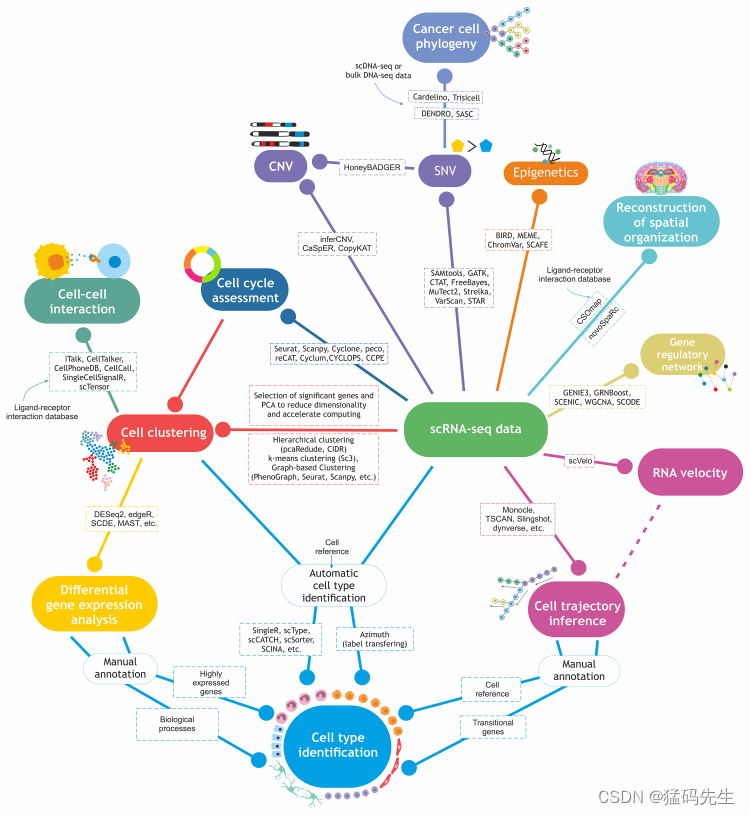
CELL CYCLE EVALUATION
1.Analysis of scRNA-seq data often considers the cell cycle phases as confounders
2.Cells in the investigated samples can be at different phases of cell cycle. Therefore, cells have different expression profiles even if they belong to the same cell type.
3.This procedure is especially advisable when no actively proliferating cells are expected in the analyzed samples. When the majority of the most variable genes are represented by the cell cycle genes, which can interfere with the identification of differentially expressed genes.(However, in some cases, e.g., upon comparison of subpopulations of dividing and non-dividing cells, the information on the cell cycle stage could be important, and this confounder should not be eliminated)
4.Tools
Seurat,Scanpy,Cyclone,peco,reCAT,Cyclum,CYCLOPS,CCPE,etc.,
在数据标归一化时去除细胞周期影响,做PCA时,就看不到细胞周期相关基因对主成分的贡献
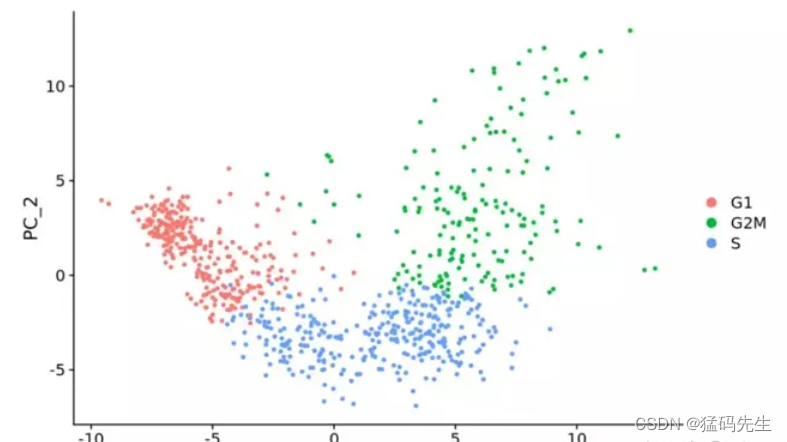 移除前后
移除前后 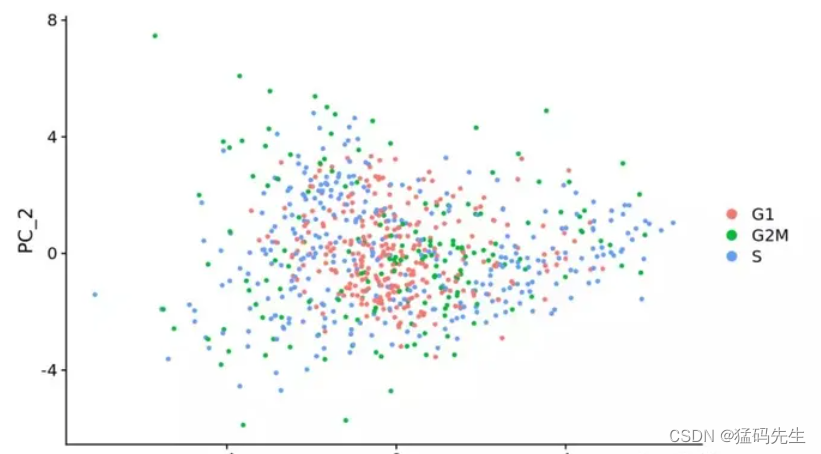
CELL CLUSTERING
hierarchical clustering
k-means clustering (SC3)
graph clustering——big scRNA-seq data
ANALYSIS OF DIFFERENTIAL GENE EXPRESSION
bulk RNA-seq data——DESeq2 and edgeR
scRNA-seq data——non-parametric Mann–Whitney test (Wilcoxon rank-sum test)provides the best results
SCDE,MAST
IDENTIFICATION OF CELL TYPES
singleR,ScType,scCATCH,scSorter,SCINA
cell clusters lacking specific markers are between the clusters with clearly pronounced markers and, hence, might represent intermediate clusters containing cells in the transitional phase between the initial and final forms.
DEVELOPMENTAL TRAJECTORIES AND RNA VELOCITY
The methods for the reconstruction of cell developmental trajectory, also called pseudotime analysis, allow to arrange the cells in the investigated sample along the modeled temporal trajectory based on the similarities of their expression profiles
Monocle,TSCAN,Slingshot
The value of RNA velocity determines the direction of the vector of each cell in the reduced-dimensionality space.
CELL–CELL COMMUNICATIONS
Development, functioning, regeneration, and homeostasis of tissues and organs are mediated by cell–cell communication, or cell–cell signaling, a process occurring through the interaction of ligands (cytokines, chemokines, hormones, growth factors, and neurotransmitters) with cell receptors.
CellPhoneDB,CellCall,SingleCellSignalR,scTensor,CellTalker,iTalk
GENE REGULATORY NETWORKS
Signalling cascades activate transcriptional factors, which interact with their binding sites in the target genes. These interactions take place in the cell nucleus and have been named gene regulatory networks.
SCENIC GENIE3 GRNBoost2
WGCNA STRING HumanNet SCODE
ANALYSIS OF CNVs
At present, many researchers define CNVs as unbalanced chromosome rearrangements – deletions and insertions of DNA sequences, whose sizes varies from several kilobases (focal) to entire chromosomes (chromosomal).
inferCNV,HoneyBADGER, Casper, CopyKAT
IDENTIFICATION OF SINGLE-NUCLEOTIDE VARIANTS
SNVs contribute to the genetic variability of living organisms, affect the course of biological processes, and could play a role of genetic factors determining predisposition to diseases.
GATK,SAMtools,GSNAP
CANCER PHYLOGENETICS
Genetic alterations, such as single-nucleotide variants and copy number aberrations, are the drivers of clonal evolution of tumor cells leading to the formation of clones and subclones resistant to antitumor therapy and having a high capacity to metastasis and recurrence.
DENDRO、Cardelino、Trisicell SASC
EPIGENOMICS: CHROMATIN ACCESSIBILITY, IDENTIFICATION OF TRANSCRIPTION FACTOR-BINDING SITES
Comparative analysis of binding motifs in the regulatory elements in combination with the information on the expression of transcription factors facilitates elucidation of the mechanisms of normal cellular processes and development of diseases.
BIRD
REPEAT ELEMENT
scTE
RECONSTRUCTION OF SPATIAL TRANSCRIPTOMICS















 344
344











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









