






参考 GitHub - davetang/defining_genomic_regions: Define regions in the genome


Exon:外显子是指基因中在mRNA剪切后保留的片段,这些片段绝大部分为编码序列。在植物和动物细胞中,大部分基因序列都被一段或几段被称为内含子的DNA序列所打断。这些基因中,那些编码蛋白质的序列被称为外显子,而那些没被表达的序列则被称为内含子
Intron:内含子又称间隔顺序,指一个基因或mRNA分子中无编码作用的片段。是真核生物细胞DNA中的间插序列。这些序列被转录在前体RNA中,经过剪接被去除,最终不存在于成熟RNA分子中。内含子和外显子的交替排列构成了割裂基因。在前体RNA中的内含子常被称作“间插序列”。在转录后的加工中,它比外显子有更多的突变。内含子是一段与编码序列无关的DNA片段,其长度一般变化很大,从几十bp至几千bp不等。
Intergenetic:基因间区域是指真核生物基因组中两个相邻基因之间的区域。这个区域可能包含一些调控序列和其他非编码DNA序列。
5'-UTR:5'-UTR是5'-非翻译区(5' Untranslated Region)的缩写,是成熟mRNA位于编码区上游不被翻译为蛋白质的区域。这个区域从mRNA的5'端起始,一直延伸到起始密码子AUG开始的地方。
3'-UTR:3'-UTR是3'-非翻译区(3'-Untranslated Region)的缩写,是成熟mRNA位于编码区下游不被翻译为蛋白质的区域。
Step1: 下载genecode上人类的基因注释文件
截至目前的下载链接
https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_44/gencode.v44.annotation.gtf.gz
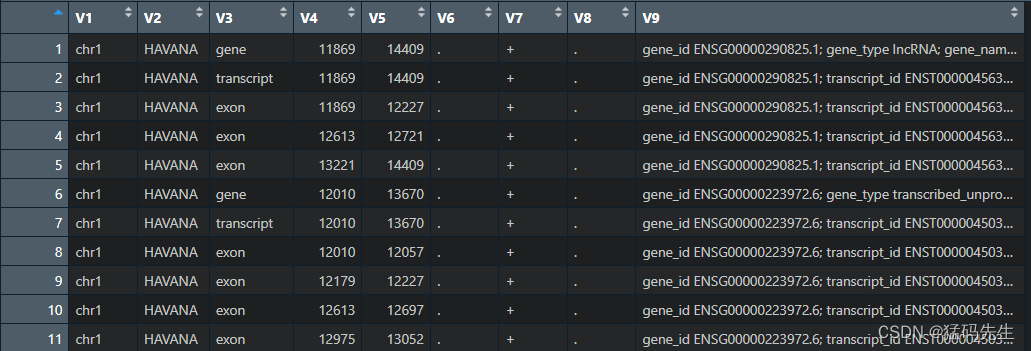
可以看到,GTF注释文件,分为了几大类
CDS exon gene Selenocysteine start_codon stop_codon transcript UTR 编码区 外显子 基因 硒代半胱氨酸 起始密码子 终止密码子 转录本 非翻译区
1.1 gene包含一个或多个transcript
1.2 每个transcript上,分为一个或多个exon
1.3 exon组成分为三种情况
exon包含5’UTR+CDS
exon包含CDS
exon包含CDS+3’UTR
1.4 起始密码子属于5’UTR+CDS中CDS区域,终止密码子不属于CDS+3’UTR中的CDS区域
Step2: 提取exon坐标
zcat gencode.v44.annotation.gtf.gz | awk 'BEGIN{OFS="\t";} $3=="exon" {print $1,$4-1,$5}' | sort -k1,1 -k2,2n| bedtools merge > hg38.exon.sort.bedtools.merge.bed
Step3: 提取intron坐标
1.提取基因坐标
zcat gencode.v44.annotation.gtf.gz | awk 'BEGIN{OFS="\t";} $3=="gene" {print $1,$4-1,$5}' | sort -k1,1V -k2,2n >hg38.gene.sort.bed
2.利用基因坐标减去外显子坐标
bedtools subtract -a hg38.gene.sort.bed -b hg38.exon.sort.bedtools.merge.bed > hg38.intron.bed
3.按照染色体顺序排序
cat hg38.intron.bed | sort -k1,1V -k2,2n >hg38.intron.sort.bed
4.合并内含子坐标
bedtools merge -i hg38.intron.sort.bed > hg38.intron.sort.bedtools.merge.bed
5.删除中间文件
rm hg38.gene.sort.bed
rm hg38.intron.bed
rm hg38.intron.sort.bed
Step4: 提取Intergenetic坐标
1.提取基因坐标
zcat gencode.v44.annotation.gtf.gz | awk 'BEGIN{OFS="\t";} $3=="gene" {print $1,$4-1,$5}' | sort -k1,1V -k2,2n >hg38.gene.sort.bed
2.生成染色体size数量文件
mysql --user=genome --host=genome-mysql.cse.ucsc.edu -A -e "select chrom, size from hg38.chromInfo" > hg38.genome
3.删除染色体size数量文件第一行
sed '1d' hg38.genome > hg38_1.genome
4.按照染色体顺序排序
cat hg38_1.genome|sort -k1,1V -k2,2n >hg38.sort.genome
5.利用bedtools complement提取基因间坐标
bedtools complement -i hg38.gene.sort.bed -g hg38.sort.genome > hg38.intergenetic.sort.bedtools.bed
6.删除中间文件
rm hg38.gene.sort.bed
rm hg38.genome
rm hg38_1.genome
Step5: 提取Intragenic坐标
暂无
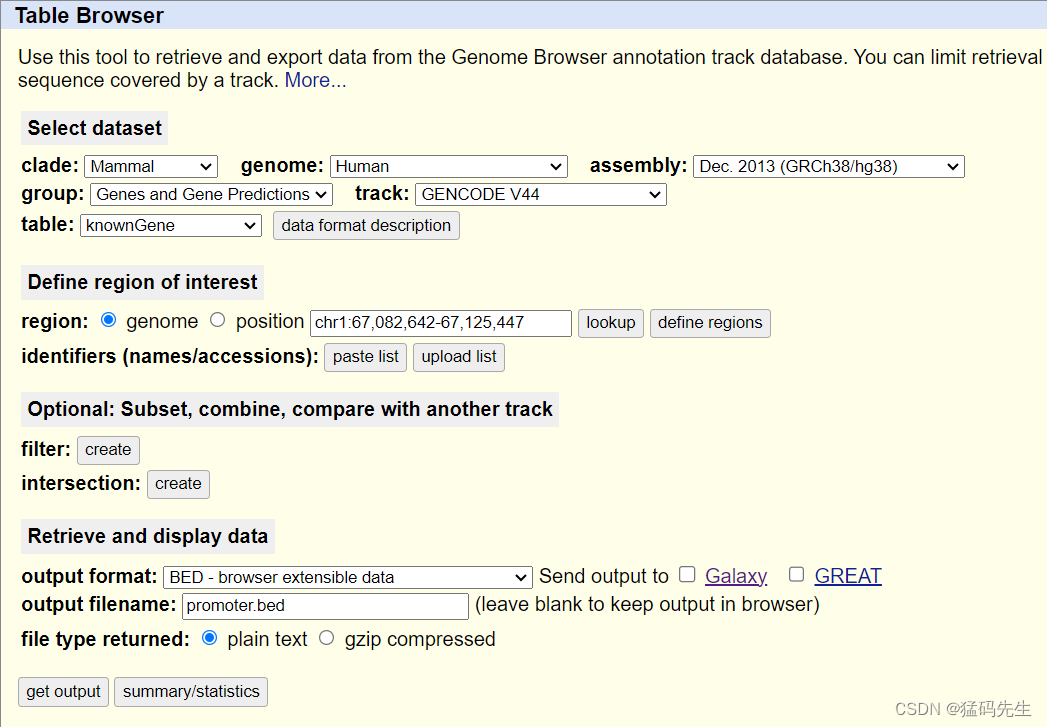
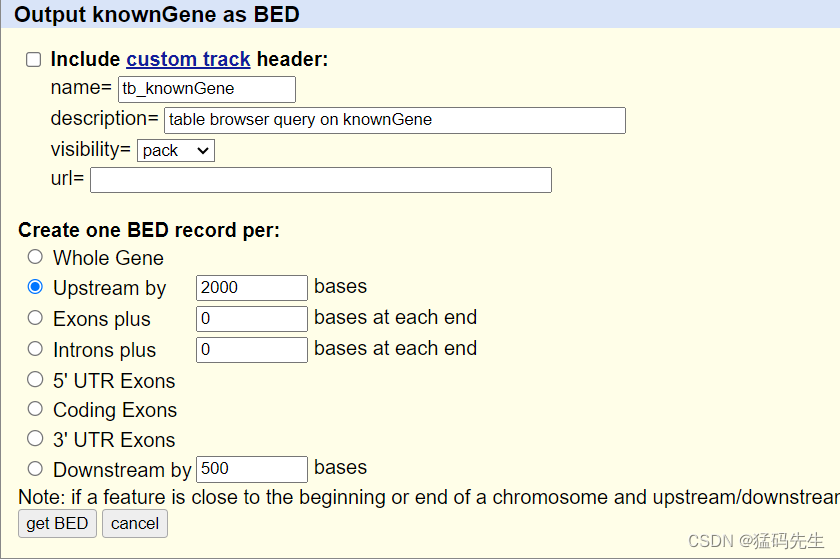
Step6: 提取Promoter坐标
在UCSC中下载,提取Tss位点上方0-2000bp的区间,如果手动的话,注意+链为TSS直接减去2000bp,-链为Tss直接加上2000bp
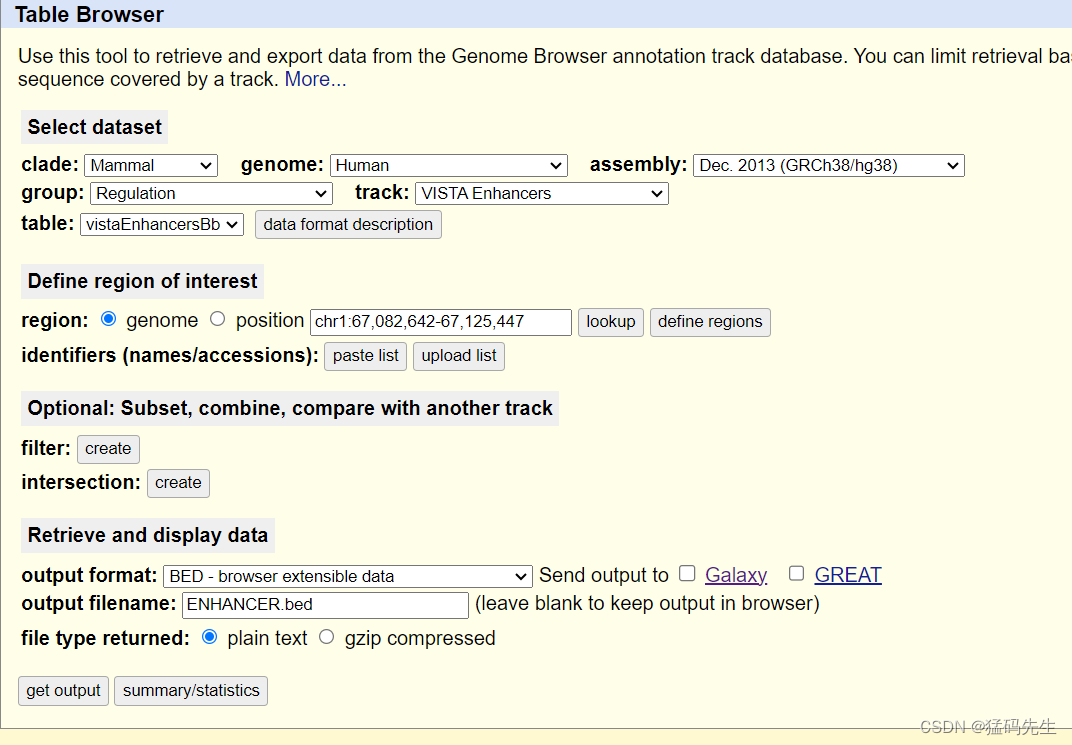
Step7: 提取Enhancer坐标
在UCSC中下载
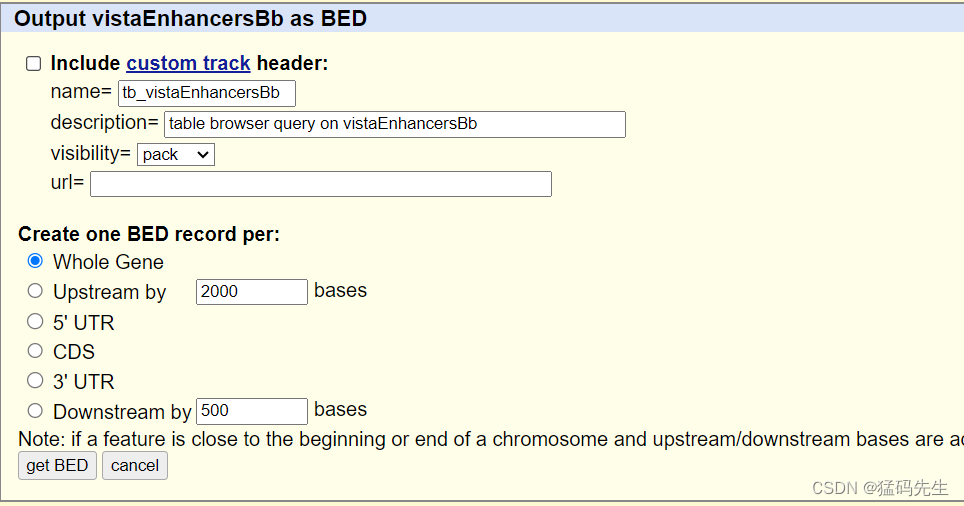
Step9: 提取CpG island坐标
在UCSC中下载
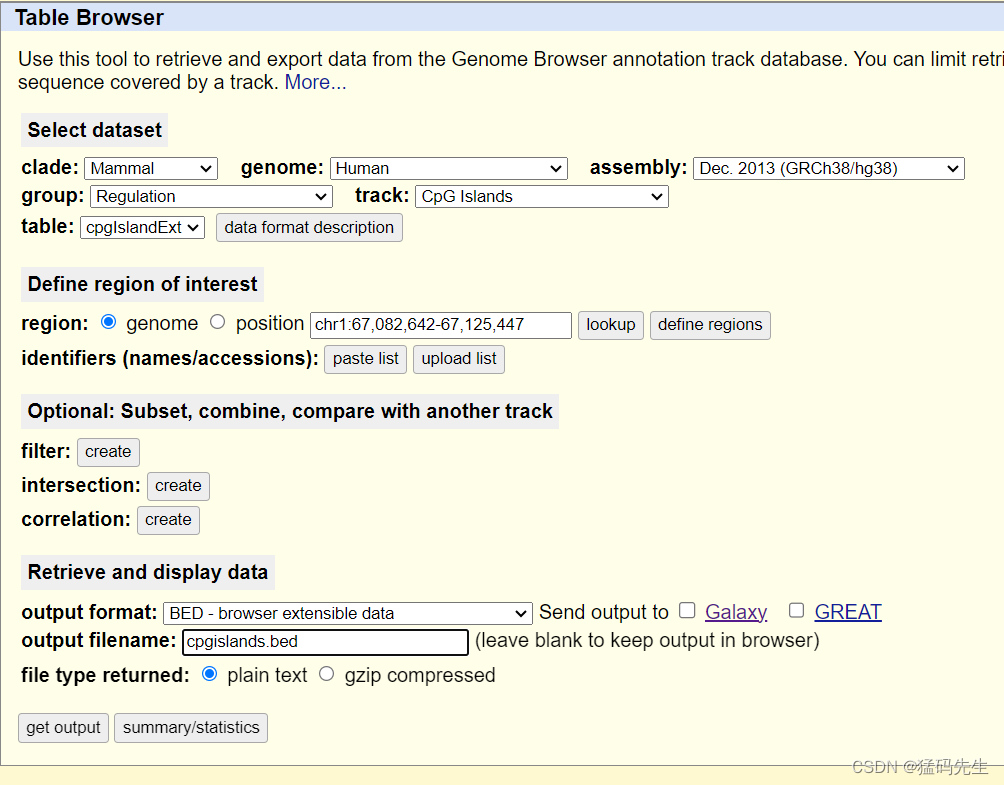
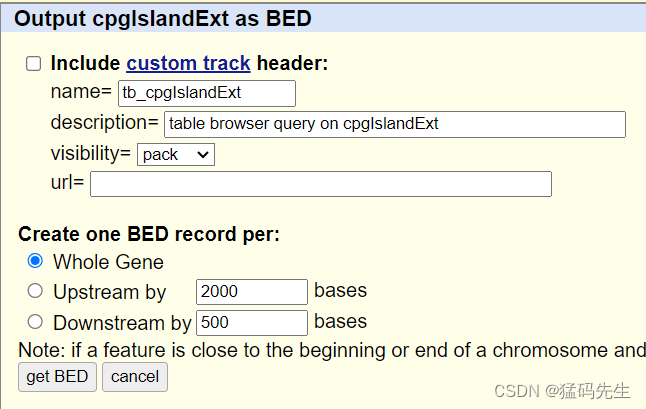
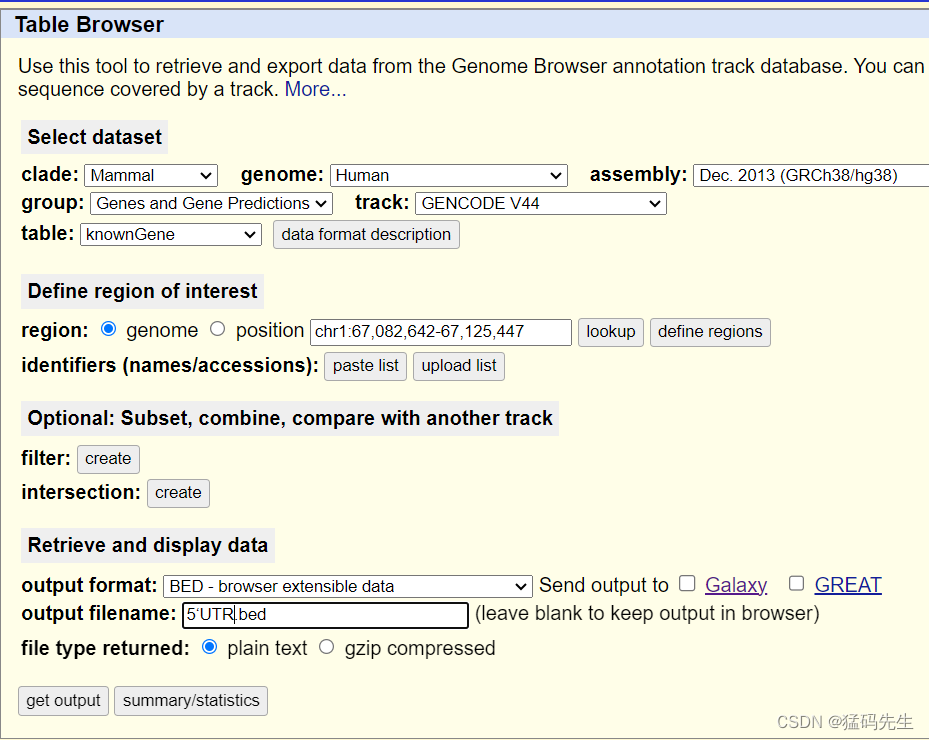
Step9: 提取5'UTR坐标
在UCSC中下载
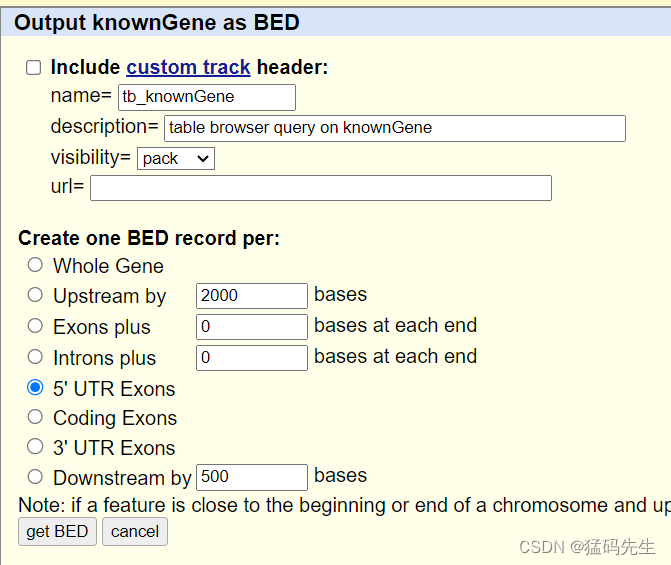
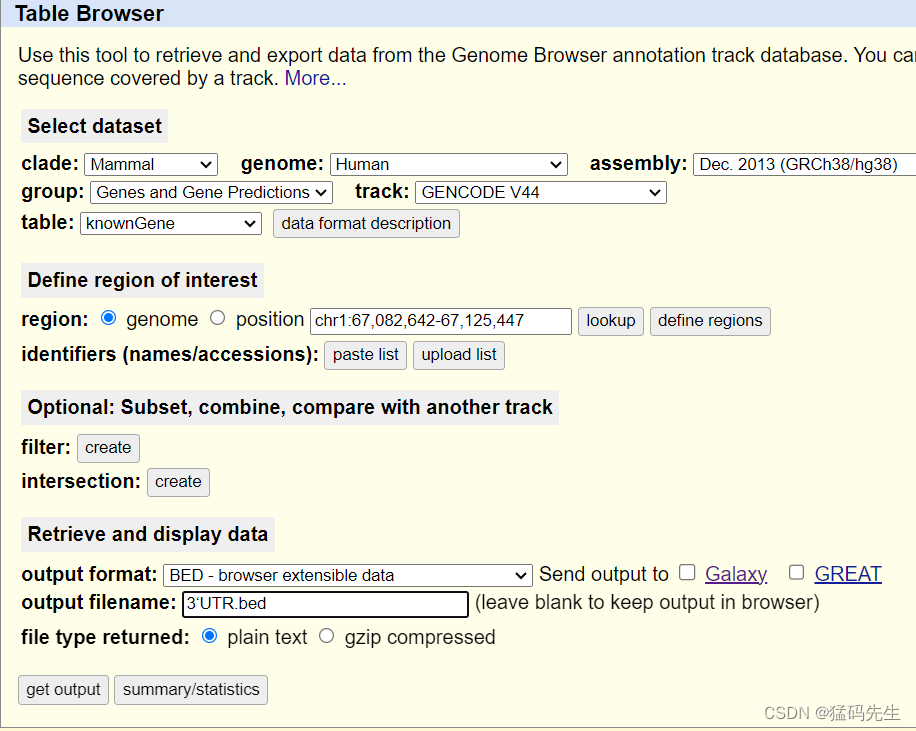
Step10: 提取3'UTR坐标
在UCSC中下载
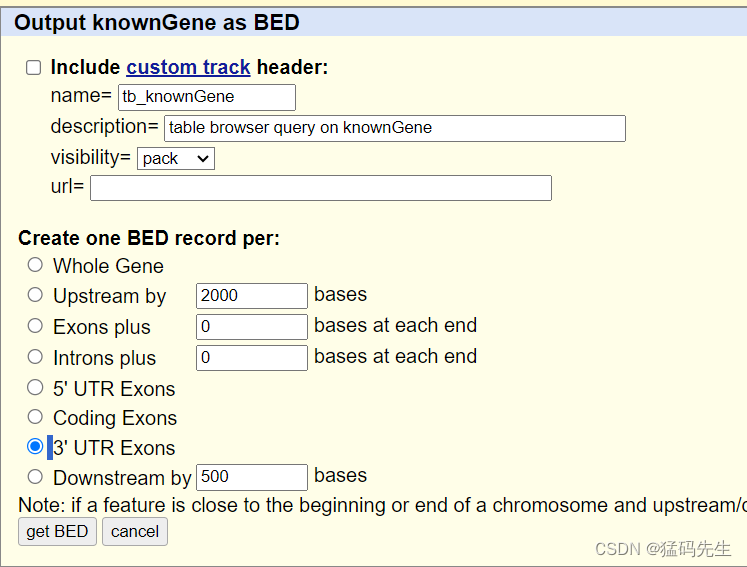
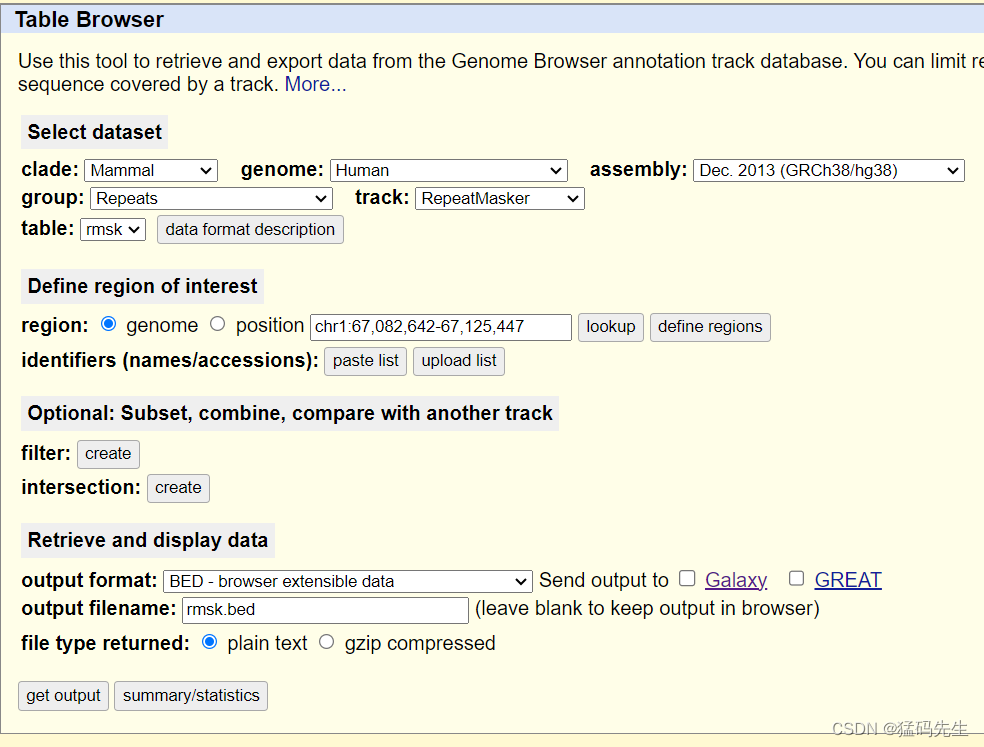
Step11: 提取repeat坐标
在UCSC中下载

















 578
578

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









