






目录
- DA-302168S,一种新型口服 GLP-1R 激动剂,展现治疗 2 型糖尿病和肥胖症的良好前景,目前已进入 II 期临床试验。
- 沉默 Hif1α、KLF4 和 SHH 基因可逆转黑色素瘤干细胞特性,为治疗提供新策略。
- 研究揭示柚子成分 DHB 是如何抑制 CYP3A4 酶活性,影响药物代谢。
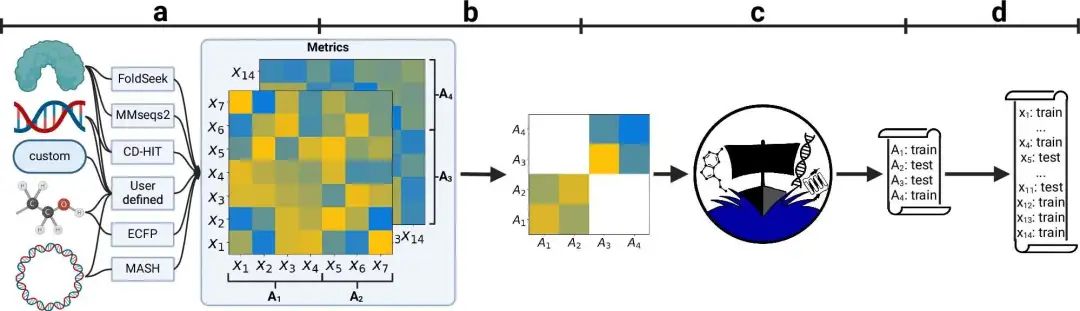
- DataSAIL 通过优化数据分割,减少相似性泄漏,提升机器学习模型在生物数据上的泛化能力。
- PAMmla 模型助力定制 CRISPR-Cas9,实现更精准、更安全的基因编辑。
- TrialGenie 利用真实世界健康数据和多智能体框架,简化并改进临床试验设计。
- 新方法利用 CT 扫描图像预测脑出血扩张,准确率超越现有临床方案。
- DNABERT-Epi 结合 DNA 序列和表观遗传特征,显著提高了 CRISPR/Cas9 脱靶效应预测的准确性。
- MetaMolGen,新的 AI 模型,用少量数据就能设计新分子,还能控制分子特性!
- Bio-DTA 模型结合 DNA 序列和单细胞转录组数据,精准预测基因调控网络。
1. 口服 GLP-1 激动剂 DA-302168S 展现 2 型糖尿病治疗潜力
2 型糖尿病和肥胖症是全球关注的健康问题,而胰高血糖素样肽 -1 受体(GLP-1R)激动剂是治疗这两类疾病的重要靶点。目前市面上虽然已有多种口服小分子 GLP-1R 激动剂,但它们普遍存在一些局限性,例如效力低、药代动力学特性差以及安全性问题。
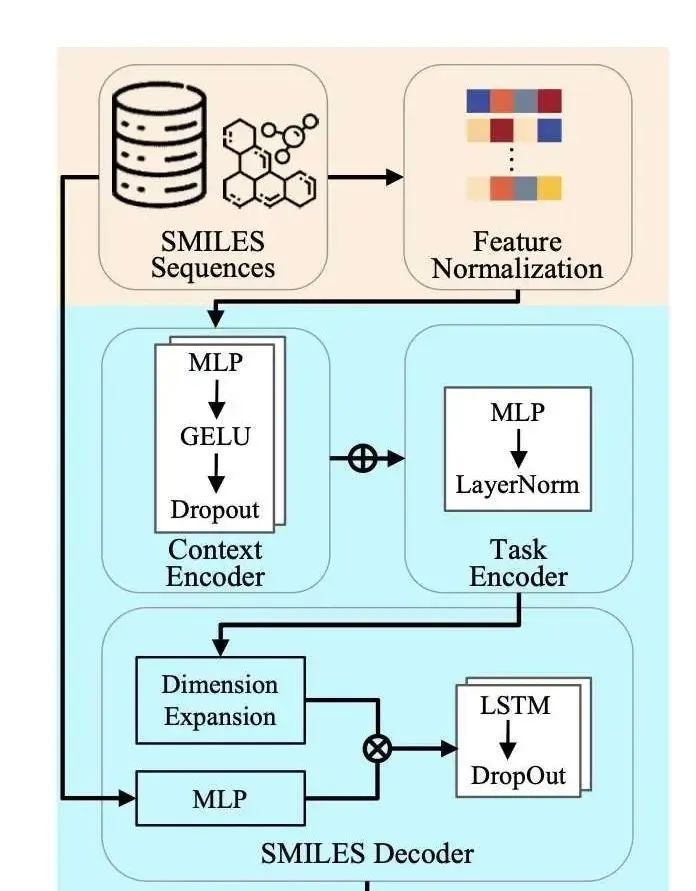
研究者开发了一种名为 DA-302168S (化合物 29) 的新型口服小分子 GLP-1R 激动剂,它展现出更高的体内外效力,并且降低了药物相互作用的风险。临床前研究数据表明,DA-302168S 在激活 cAMP、降低血糖和抑制食欲方面均表现出显著疗效。安全性评估显示,DA-302168S 的 hERG IC50 > 30 μM,脱靶毒性低,安全性良好。此外,它还具有良好的药代动力学特性,支持每日一次的口服给药,从而提高患者的依从性。
DA-302168S 已成功完成 I 期临床试验,目前正在进行 II 期临床试验。这些研究结果表明,DA-302168S 有望成为治疗 2 型糖尿病和肥胖症的一种极具潜力的疗法。
2. 抑制基因表达逆转黑色素瘤干细胞特性
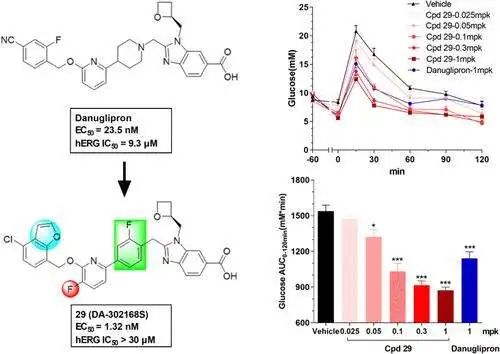
恶性黑色素瘤是一种高度侵袭性的皮肤癌,其部分驱动因素是具有显著自我更新、分化和治疗抵抗能力的癌症干细胞 (CSCs)。这项研究探讨了沉默三个关键基因——Hif1α、KLF4 和 SHH——如何影响黑色素瘤 CSCs 的特性。
研究人员利用基于小干扰 RNA (siRNA) 的方法,观察到基因和蛋白质水平的显著变化,揭示了这些通路如何影响黑色素瘤进展。结果表明,沉默这些基因降低了 CSCs 的干细胞样特征。值得一提的是,沉默 Hif1α 触发了缺氧相关基因表达的显著下降,而靶向 SHH 导致 Gli1(SHH 信号的下游效应器)表达减少,突出了其作为治疗靶点的潜力。
研究人员还观察到表观遗传标记(如 HDAC9 和 EP300)的变化,这些标记在维持干性和调节基因表达中起着至关重要的作用。有趣的是,这些干预似乎重编程了 CSCs,促使它们转向一种不同于传统 CSCs 和非干细胞癌细胞 (NCSCs) 的表型。
3. 柚子成分如何影响药物代谢?CYP3A4 的秘密
柚子可能对某些药物的代谢产生意想不到的影响。这背后的秘密在于一种名为 6’,7’-二羟基佛手柑内酯(DHB)的天然呋喃香豆素,它能够引起包括 CYP3A4 在内的人体多种细胞色素 P450 酶(P450s)的机制性失活(MBI)。CYP3A4 可是人体内重要的药物代谢酶,它的失活会导致药物在体内蓄积,增加不良反应的风险。
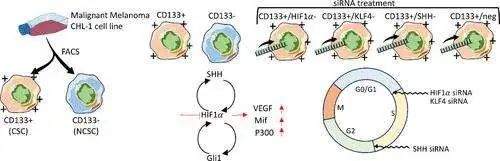
为了揭示 DHB 影响 CYP3A4 的具体机制,研究者们结合了分子对接、分子动力学模拟和量子力学/分子力学(QM/MM)计算等多种方法。首先,他们通过分子对接和分子动力学模拟确定了 DHB 在 CYP3A4 中的结合构象。接着,QM/MM 计算模拟了两种可能的反应路径(A 和 B)。路径 A 是复合物 I(Cpd I)攻击呋喃环的 C5 位,生成 γ-酮烯醛;而路径 B 则是攻击 C4 位,生成环氧化物。
结果发现,路径 A 的活化能垒远低于路径 B,这意味着生成 γ-酮烯醛的反应在动力学上更有优势。而且,γ-酮烯醛的热力学稳定性也高于环氧化物。换句话说,即使最初生成了环氧化物,它也很可能在酶内或水溶液中重排成 γ-酮烯醛。
研究者们得出结论:γ-酮烯醛是 DHB 被 CYP3A4 氧化后的唯一最终产物。这项研究不仅深入解析了柚子成分对 CYP3A4 失活的影响机制,也为我们理解食物与药物的相互作用提供了宝贵的线索。这也提醒我们,看似平常的食物也可能对药物代谢产生影响,在服用某些药物时,需要注意饮食搭配,避免潜在的风险。
4. DataSAIL:让生物数据机器学习更可靠
在生物信息学领域,传统的随机数据分割方法常常因为分子间的相似性导致信息泄漏,从而影响模型评估,使得像 ROC-AUC 和 RMSE 等指标虚高。DataSAIL 这款 Python 工具包,将数据分割问题转化为 NP-hard 优化问题,通过最小化训练集和测试集之间的相似性来解决信息泄漏,从而更好地评估模型在预测分子毒性或药物 - 靶点相互作用等方面的泛化能力。这对于确保模型不仅能记住训练数据,还能泛化到新的生物学场景至关重要。
研究人员提出了 (k, R, C)-DataSAIL 问题,通过最小化训练集和测试集之间的总相似性 L(π),优化一维或二维数据集的折叠分配。他们引入了一种使用聚类和整数线性规划 (ILP) 的启发式算法,可以在蛋白质、小分子等数据上进行可扩展的分层和相似性感知的分割。该框架支持自定义相似性函数(例如,分子的 Tanimoto,蛋白质的 MMseqs2)和分层类别平衡,这在类别成员(如毒性)与特征相似性一致时至关重要。
研究人员使用五个机器学习模型(RF、SVM、XGB、MLP 和 D-MPNN 或 DeepDTA)在 Tox21、QM8 和 LP-PDBBind 等一维和二维数据集上进行了评估。评估指标包括 MAE、RMSE 和 ROC-AUC,以及自定义泄漏指标 L(π)。DataSAIL 始终生成泄漏最低的分割,并揭示了与随机分割相比更明显的性能下降,这突出了它在揭示真实泛化能力方面的有效性。例如,D-MPNN 的性能在 DataSAIL 的相似性感知分割上显著下降,这表明依赖标准评估中相似性引起的捷径存在风险。
从技术上讲,DataSAIL 允许多类型实体分割(例如,药物 - 蛋白质对),支持自定义相似性矩阵,并使用 GUROBI、MOSEK 或 SCIP 求解器。从生物学角度来看,它有助于在推理数据与训练数据不同的情况下进行严格的模型验证,例如在药物发现流程中。这确保了模型不仅在熟悉的分子骨架上进行评估,还在新的化学和遗传结构上进行评估,最终促进了生物医学研究中稳健的、具有转化意义的 AI 模型的开发。
📜Paper: https://www.nature.com/articles/s41467-025-58606-8
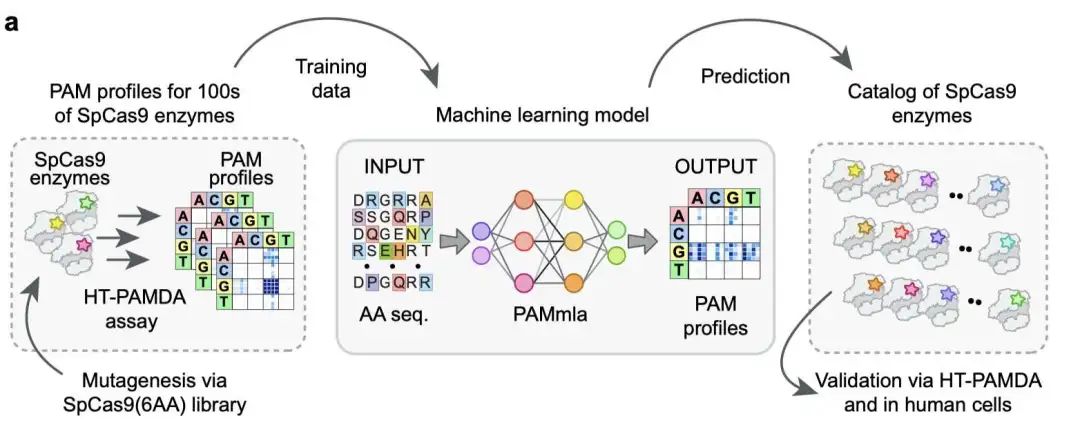
5. 定制 CRISPR:精准基因编辑新时代
这项发表于《自然》杂志的研究介绍了一种名为 PAMmla 的机器学习模型。这个模型可以预测数百万种 SpCas9 变体的 PAM 特异性。换句话说,它能帮助科学家设计定制化的 Cas9 酶,实现精准的基因靶向,减少脱靶效应。这比传统的广谱酶更精准、更安全。
研究者结合了高通量蛋白工程、饱和诱变和 PAMmla 模型,构建了一个包含 6400 万个 SpCas9 变体的库。他们实验验证了近 1000 种酶,并通过计算预测了整个库的 PAM 特异性。
不同于传统的 PAM 松弛 Cas9 变体(例如 SpG、SpRY),PAMmla 允许创建高度特异性的 PAM 改变酶。这些酶在保持高编辑效率的同时,最大限度地减少了脱靶切割。
利用 PAMmla 指导的计算机模拟定向进化 (ISDE) 方法,研究团队成功设计了用于等位基因特异性编辑的 Cas9 酶。例如,他们能够区分野生型和突变型等位基因,这在视网膜色素变性等疾病(例如 RHO P23H 突变)中至关重要。
PAMmla 预测的酶在人体细胞和体内小鼠模型中均表现出优异的性能,实现了更高的编辑效率,并显著降低了脱靶效应(与 SpRY 相比,脱靶位点减少高达 96%),并通过 GUIDE-seq-2 测定得到验证。
在治疗应用方面,PAMmla 设计的酶能够对患者来源细胞中的 CYBB T362I 突变进行等位基因选择性校正,以治疗 X 连锁慢性肉芽肿病,实现了 >90% 的校正率,且脱靶编辑极少。
这项研究扩展了 Cas9 酶的工具包,引入了能够靶向非经典 PAM(例如 NGAT、NGTC、NGCM)的 PAM 改变变体,提供了比广谱 PAM 松弛变体更广泛、更安全的基因组编辑选择。
结构分析和 SHAP 解释性方法证实了 PAM 选择性的机制基础,揭示了特定氨基酸取代如何调节与靶 DNA 的碱基特异性或骨架相互作用。
这项工作突出了实验蛋白工程与机器学习之间的协同作用,为快速、可定制的 Cas9 变体开发提供了一个可扩展的框架,以满足不同的基因组编辑需求。
研究者提供了一个交互式网络工具和 6400 万个酶库的所有 PAMmla 预测结果,供其他研究人员访问和设计具有所需 PAM 特异性的 SpCas9 变体。
6. AI 智能化临床试验设计:TrialGenie
TrialGenie 是一个多智能体框架,旨在利用真实世界健康数据加速临床试验设计。系统集成了五个专业的 AI 智能体:主管、试验员、信息学家、临床医生和统计学家。每个智能体都由大型语言模型驱动,并具备特定领域能力,通过结构化对话协作,从电子健康记录中提取洞见,从而改进临床试验设计。
传统的临床试验设计耗时且需要大量的专业知识,而来自电子健康记录的真实世界数据包含可用于指导此过程的宝贵证据。虽然已开发出机器学习方法来提取真实世界证据,但它们仍然需要与领域专家进行大量沟通。TrialGenie 通过创建一个智能体框架来解决这个问题,该框架通过专业智能体之间的迭代对话和分析来自主优化试验方案。
TrialGenie 架构包括五个具有特定角色的智能体:主管充当中央决策者;试验员检索并标准化试验信息;信息学家将试验信息映射到电子健康记录数据;临床医生提供领域专业知识;统计学家进行分析。这些智能体使用 Trial Retriever、RAG 和 Trial Simulator 等工具协同开发试验方案、提取相关数据、进行统计分析并生成综合报告。
该系统使用四种大型语言模型(GPT-4o、Phi-4、DeepSeek-R1 和 Gemma 3)在多项任务中进行了评估。GPT-4o 的表现始终优于其他模型,在实体解析中实现了 98.6% 的召回率和 92.3% 的精确率,SQL 生成错误最少(总共 12 个),临床推理得分最高(平均 4.66/5),并准确估计了接近真实值的风险比。该系统通过三个疾病特异性案例研究(用于心力衰竭的奈西利肽、用于肾损伤的肾脏替代疗法和用于脓毒性休克的氢化可的松)进行了演示,展示了其生成全面试验模拟方案和统计分析的能力。
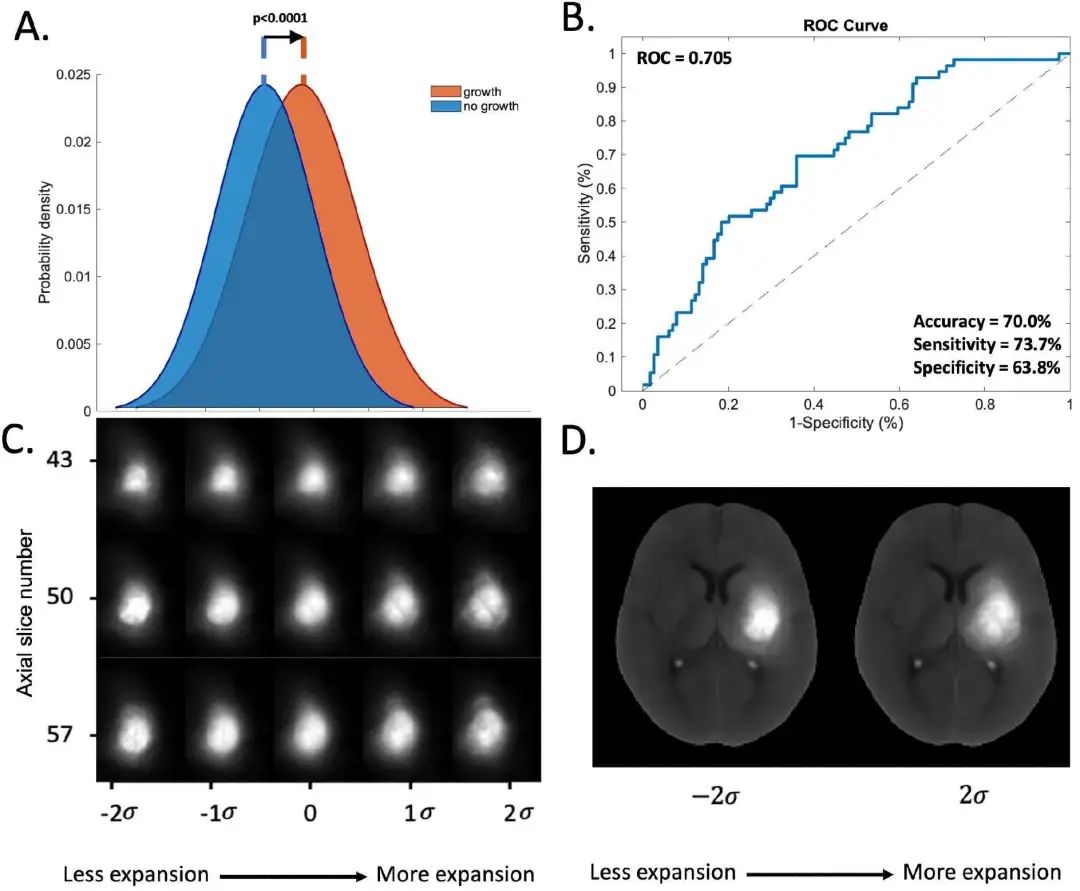
7. AI 预测脑出血扩张,准确率达 71%
脑出血 (ICH) 患者的血肿扩张是导致不良后果和死亡的主要因素。早期预测血肿扩张对于及时干预至关重要。弗吉尼亚大学的研究人员开发了一种名为基于传输形态学 (TBM) 的新方法,使用标准的非对比增强计算机断层扫描 (NCCT) 图像预测 ICH 患者的血肿扩张。
研究人员使用来自 VISTA-ICH 数据库的 170 名患者进行模型开发,并使用来自 ERICH 研究的 170 名患者进行外部验证。结果显示,TBM 预测 24 小时血肿扩张风险的 AUROC 为 0.71,优于现有的临床方案和其他机器学习方法,如支持向量机 (SVM)、逻辑回归、k 近邻和 3D ResNet CNN。
TBM 模型识别出与血肿扩张相关的四个关键形态特征:体积较大、密度异质性、形状不规则和外周密度分布。血肿的位置也被发现是扩张的独立预测因子,朝向丘脑、内囊后肢和侧脑室房的血肿显示出更高的扩张率。
这项研究的创新之处在于其兼具预测能力和可解释性。TBM 不仅可以预测血肿扩张,还可以帮助研究人员可视化和理解与扩张相关的形态特征。这些特征与先前提出的生物学机制(如“雪崩效应”)相符,其中出血由于相邻血管的机械剪切而扩张。
通过检测 NCCT 图像中的细微模式,TBM 为 ICH 患者的早期风险分层和干预提供了潜力,而无需专门的成像技术。这可以加快治疗决策,特别是随着手术清除技术的最新进展,并通过更好地了解扩张机制来帮助开发预防性疗法。
📜Paper: https://elifesciences.org/reviewed-preprints/105782
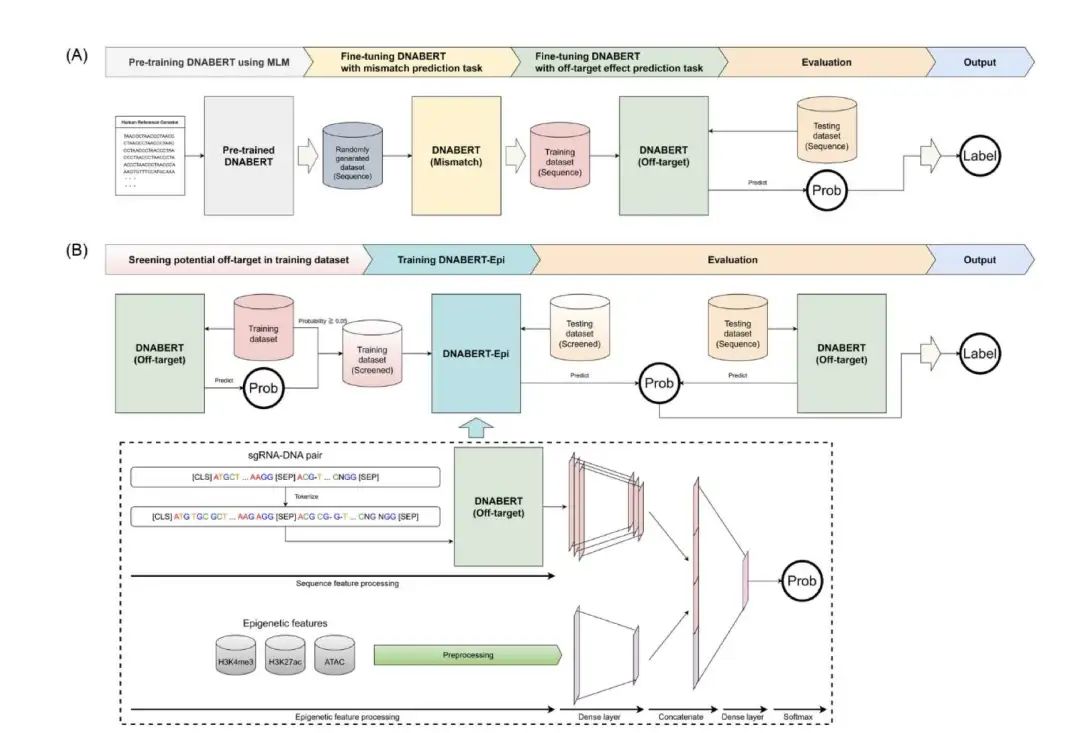
8. DNABERT-Epi 预测 CRISPR 脱靶效应更精准
DNABERT-Epi 是一种新的 CRISPR/Cas9 脱靶效应预测方法。它结合了 DNABERT(一种预训练的 DNA 语言模型)和表观遗传特征,例如染色质可及性数据(H3K4me3、H3K27ac 和 ATAC-seq)。这种结合使模型能够更好地理解 Cas9 活性,从而更准确地预测体内脱靶位点。
研究者分两个阶段微调了 DNABERT:首先进行错配预测任务以模拟 sgRNA-DNA 配对,然后进行二元分类以预测脱靶切割。这种方法使模型能够学习生物学相关的序列相互作用。表观遗传数据通过一个专用的前馈网络整合到 DNABERT-Epi 中,实现了序列和染色质环境的整合,尤其提升了在 GUIDE-seq 数据(反映细胞内条件)上的性能。
DNABERT-Epi 在 F1、MCC、ROC-AUC 和 PR-AUC 等关键指标上均优于五种现有模型(GRU-Emb、CRISPR-BERT、CRISPR-HW、CRISPR-DIPOFF 和 CrisprBERT),尤其是在对不平衡数据集至关重要的精确率 - 召回率指标上表现出色。消融研究证实了 DNABERT 预训练的必要性;从头开始训练的模型无法学习有意义的模式,这突出了大规模基因组预训练对于 CRISPR 预测任务的重要性。进一步的消融实验表明,整合表观遗传特征比仅使用序列的模型提供了统计学上显著的改进,验证了染色质环境在脱靶预测中的生物学相关性。
该研究使用了来自 CHANGE-seq 和 GUIDE-seq 的高通量数据集,以确保稳健的评估,包括跨实验平台的迁移学习场景。将 DNABERT-Epi 与基线模型相结合的集成方法进一步提高了预测的稳健性,利用了不同架构的互补优势。DNABERT 的注意力权重可视化显示,该模型在预测过程中优先考虑种子区域(Cas9 特异性的关键),为了解其决策过程提供了见解,并与已知的生物学机制相符。
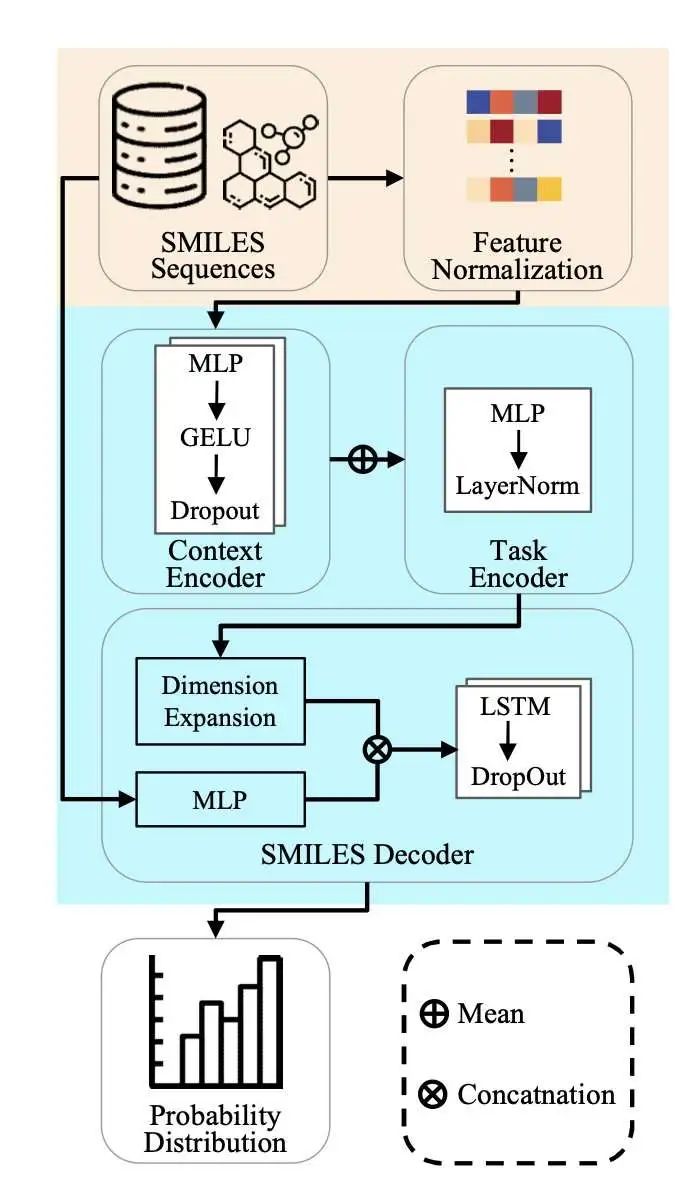
9. 快速分子设计:MetaMolGen 来了!
MetaMolGen 是一个基于元学习的分子生成模型,特别擅长在少量数据和特定属性条件下设计新分子。与传统的基于图或序列的分子生成器不同,MetaMolGen 结合了条件神经过程(CNPs)和 Reptile 元学习算法,只需少量数据就能快速适应新的分子设计任务。
它通过将分子特征投影到标准化潜在空间来标准化图基序分布,并使用基于 LSTM 的自回归解码器生成 SMILES 序列,确保化学有效性和结构多样性。为应对数据稀缺的情况,MetaMolGen 利用可学习的标准化模块,稳定特征分布,增强梯度流,并减少训练不稳定性,这在孤儿药发现等资源匮乏的环境中尤为重要。
该框架通过属性投影仪支持特定属性的分子生成,允许生成具有所需物理化学属性(例如 LogP、TPSA、QED)的分子,确保与目标属性一致。在多个基准测试(ChEMBL、QM9、ZINC、MOSES)中,MetaMolGen 在独特性、药物相似性、可合成性和溶解性方面优于 MolGAN、ORGAN、RNN 和 MolGPT 等基线模型,同时保持了竞争性的有效性。
虽然有效性略低于 MolGAN(75% vs. 97%),但 MetaMolGen 实现了更高的独特性(约 99.9%)、多样性和药物相关特性,证明了在实际药物设计至关重要的多个目标之间取得了更好的平衡。在针对真实药物(阿司匹林、达菲、阿莫西林、氯喹)的特性进行条件生成实验中,MetaMolGen 成功生成了与目标属性一致的多种分子,展示了强大的可控性。
该模型的少量样本学习能力允许从只有 1000 个分子的数据集中进行有效学习,性能可扩展到 10000 个样本,即使在有限的监督下也能保持高有效性和属性控制。MetaMolGen 的架构结合了特征归一化、元学习和自回归解码,为分子设计提供了一个通用的、数据高效的解决方案,在药物发现、材料科学等领域具有潜在的应用价值。
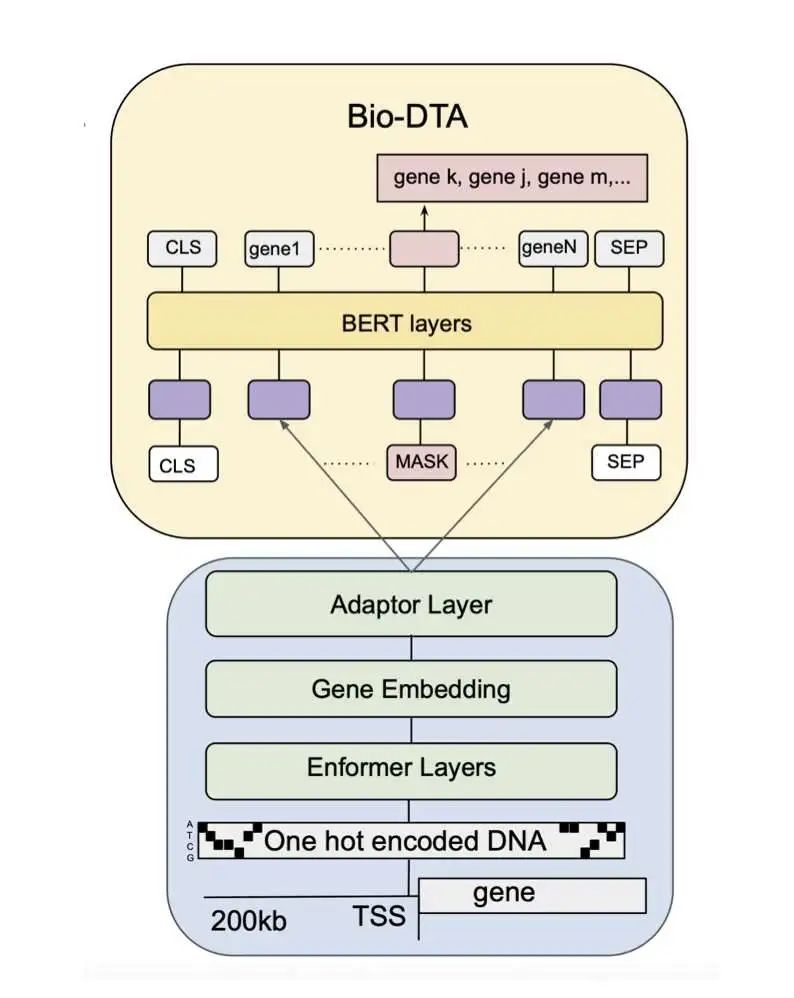
10. Bio-DTA:动态基因表达模型
Bio-DTA 是一项创新性的研究成果,它将 DNA 序列数据与单细胞转录组结合起来,构建了一个全新的多模态基础模型。通过动态标记适配 (DTA),Bio-DTA 能够捕捉基因变异如何影响单细胞水平的基因表达。不同于以往将基因标记视为固定实体,Bio-DTA 用 DNA 序列嵌入的投影取代了单细胞模型中静态的标记嵌入,使基因表征能够动态反映潜在的基因组序列。
实验证明,Bio-DTA 在预测基因调控网络方面优于传统的单模态模型(如 Geneformer),它对基因扰动及其下游转录效应更为敏感。例如,在胎儿心肌细胞中对转录因子 GATA4 启动子进行计算机模拟诱变,Bio-DTA 准确捕捉到了 GATA4 直接和共同结合靶点(如 TBX5)上下文嵌入的变化,其表现优于 Geneformer 和无适配器基线。
此外,Bio-DTA 还能学习检测共调控动态。在 GATA4 转录起始位点附近的随机突变,导致其经过实验验证的靶基因的嵌入发生显著变化,而管家基因保持稳定,这突出了模型的生物学特异性。与基于删除的方法(关键基因标记会从输入序列中完全移除)相比,动态标记适配方法使 Bio-DTA 能够在不改变全局输入的情况下响应细微的基因变化。
Bio-DTA 利用双向 Transformer 编码器架构,在超过 3300 万个单细胞转录组以及来自 Ensembl GRCh38 参考的基因组序列数据上进行训练,并通过 Enformer 提取序列嵌入。这项研究表明,将 DNA 序列上下文与转录组模型相结合,丰富了单细胞嵌入的可解释性和生物学相关性,为在不同遗传背景下模拟基因调控开辟了新途径。未来,研究者计划将 Bio-DTA 的应用扩展到其他转录因子、细胞类型和更复杂的遗传变异(如 SNP、插入缺失),并整合 RNA 异构体和蛋白质序列嵌入,以获得更丰富的多模态表征。通过连接 DNA 序列和单细胞表达数据,Bio-DTA 提供了一个灵活的框架,用于理解单细胞分辨率下的基因调控,并在个性化医疗、调控基因组学和系统生物学中具有潜在应用价值。
如何学习大模型 AI ?
由于新岗位的生产效率,要优于被取代岗位的生产效率,所以实际上整个社会的生产效率是提升的。
但是具体到个人,只能说是:
“最先掌握AI的人,将会比较晚掌握AI的人有竞争优势”。
这句话,放在计算机、互联网、移动互联网的开局时期,都是一样的道理。
我在一线互联网企业工作十余年里,指导过不少同行后辈。帮助很多人得到了学习和成长。
我意识到有很多经验和知识值得分享给大家,也可以通过我们的能力和经验解答大家在人工智能学习中的很多困惑,所以在工作繁忙的情况下还是坚持各种整理和分享。但苦于知识传播途径有限,很多互联网行业朋友无法获得正确的资料得到学习提升,故此将并将重要的AI大模型资料包括AI大模型入门学习思维导图、精品AI大模型学习书籍手册、视频教程、实战学习等录播视频免费分享出来。

第一阶段(10天):初阶应用
该阶段让大家对大模型 AI有一个最前沿的认识,对大模型 AI 的理解超过 95% 的人,可以在相关讨论时发表高级、不跟风、又接地气的见解,别人只会和 AI 聊天,而你能调教 AI,并能用代码将大模型和业务衔接。
- 大模型 AI 能干什么?
- 大模型是怎样获得「智能」的?
- 用好 AI 的核心心法
- 大模型应用业务架构
- 大模型应用技术架构
- 代码示例:向 GPT-3.5 灌入新知识
- 提示工程的意义和核心思想
- Prompt 典型构成
- 指令调优方法论
- 思维链和思维树
- Prompt 攻击和防范
- …
第二阶段(30天):高阶应用
该阶段我们正式进入大模型 AI 进阶实战学习,学会构造私有知识库,扩展 AI 的能力。快速开发一个完整的基于 agent 对话机器人。掌握功能最强的大模型开发框架,抓住最新的技术进展,适合 Python 和 JavaScript 程序员。
- 为什么要做 RAG
- 搭建一个简单的 ChatPDF
- 检索的基础概念
- 什么是向量表示(Embeddings)
- 向量数据库与向量检索
- 基于向量检索的 RAG
- 搭建 RAG 系统的扩展知识
- 混合检索与 RAG-Fusion 简介
- 向量模型本地部署
- …
第三阶段(30天):模型训练
恭喜你,如果学到这里,你基本可以找到一份大模型 AI相关的工作,自己也能训练 GPT 了!通过微调,训练自己的垂直大模型,能独立训练开源多模态大模型,掌握更多技术方案。
到此为止,大概2个月的时间。你已经成为了一名“AI小子”。那么你还想往下探索吗?
- 为什么要做 RAG
- 什么是模型
- 什么是模型训练
- 求解器 & 损失函数简介
- 小实验2:手写一个简单的神经网络并训练它
- 什么是训练/预训练/微调/轻量化微调
- Transformer结构简介
- 轻量化微调
- 实验数据集的构建
- …
第四阶段(20天):商业闭环
对全球大模型从性能、吞吐量、成本等方面有一定的认知,可以在云端和本地等多种环境下部署大模型,找到适合自己的项目/创业方向,做一名被 AI 武装的产品经理。
- 硬件选型
- 带你了解全球大模型
- 使用国产大模型服务
- 搭建 OpenAI 代理
- 热身:基于阿里云 PAI 部署 Stable Diffusion
- 在本地计算机运行大模型
- 大模型的私有化部署
- 基于 vLLM 部署大模型
- 案例:如何优雅地在阿里云私有部署开源大模型
- 部署一套开源 LLM 项目
- 内容安全
- 互联网信息服务算法备案
- …
学习是一个过程,只要学习就会有挑战。天道酬勤,你越努力,就会成为越优秀的自己。
如果你能在15天内完成所有的任务,那你堪称天才。然而,如果你能完成 60-70% 的内容,你就已经开始具备成为一名大模型 AI 的正确特征了。
这份完整版的大模型 AI 学习资料已经上传CSDN,朋友们如果需要可以微信扫描下方CSDN官方认证二维码免费领取【保证100%免费】


















 360
360

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









