







1、背景
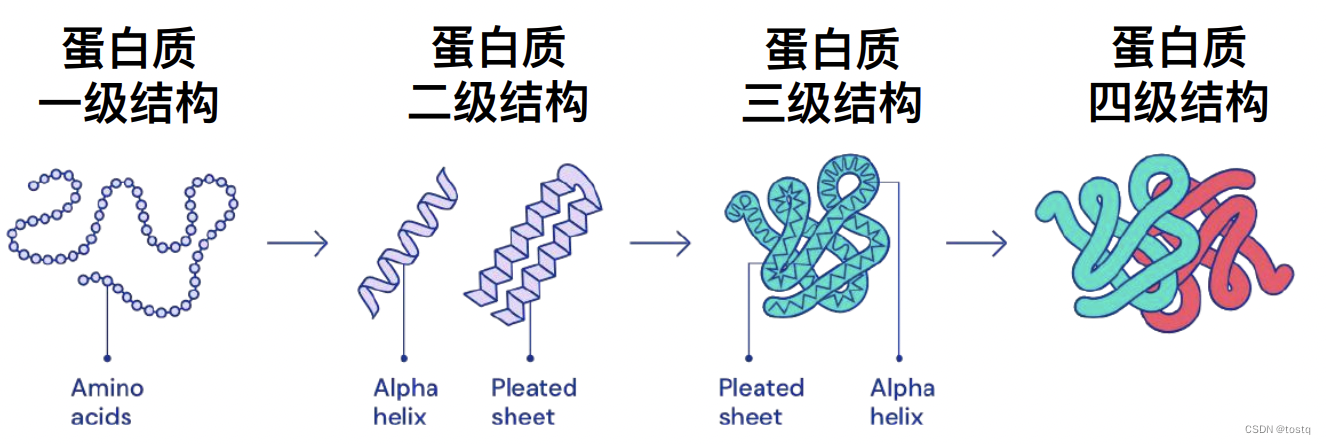
蛋白质是生物体内一类重要的生物大分子,其结构复杂多样,蛋白质的结构对于理解其功能和参与的生物学过程具有重要意义。从生物学角度上看,蛋白质的结构可以分为四个层次:初级结构、二级结构、三级结构和四级结构。
-
初级结构:初级结构是蛋白质的氨基酸序列,即蛋白质由多个氨基酸按照特定的顺序连接而成。氨基酸是蛋白质的基本组成单位,共有20种常见氨基酸,它们的不同排列形成了蛋白质的初级结构。
-
二级结构:二级结构是蛋白质中局部区域的空间排列方式。常见的二级结构包括α-螺旋和β-折叠。α-螺旋是由多个氨基酸沿着螺旋状排列而成,而β-折叠是由氨基酸链的不同区域之间的相互作用形成的折叠结构。
-
三级结构:三级结构是整个蛋白质分子的空间结构。这个结构由蛋白质的二级结构在整个分子中的排列和折叠所决定。各种相互作用力,如氢键、离子键、范德华力和疏水效应等,参与了蛋白质分子的三级结构的稳定。
-
四级结构:四级结构是由多个蛋白质分子相互组合而成的复合体,通常称为蛋白质的多聚体。多个蛋白质分子通过各种力和作用结合在一起,形成功能更为复杂的生物大分子。
其中初级结构一般可以通过Edman降解法和质谱法进行确定,而二级结构及三级结构通常需要如X射线晶体学、电镜等高度专业的设备和技术来确定,其需要极高人工及金钱成本。而AlphaFold通过深度学习技术解决蛋白质的二级结构及三级结构的预测问题,极大地提高了蛋白质结构解析效率,因此也被认为是诺奖级跨时代的工作。
2、特征输入
前文我们提到,AlphaFold根据深度学习技术,通过输入蛋白质一级结构来解析二级结构及三级结构,而一级结构为一组蛋白质氨基酸序列,但是AlphaFold的特征并不只是输入蛋白质氨基酸序列,而包含了多种相关信息。
-
单氨基酸序列target_feat
:其中f为21维氨基酸的o
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1342
1342

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









