







转录组上游分析-1
作业:
-
取出fastq文件中的所有序列ID(第一行)
less SRR1039510_1.fastq.gz | awk '{if(NR%4==1){print $0}}' less SRR1039510_1.fastq.gz | paste - - - - | cut -f 1 -
取出fastq文件中的所有序列(第二行)同上
-
对序列出现次数进行统计
less SRR1039510_1.fastq.gz | paste - - - - | cut -f 2 | sort | uniq -c | sort -nk 1如果某一序列出现次数非常高,考虑可能是核糖体RNA?或实验过程中的异常情况,可以比对序列来查看该序列来源
数据质控
数据质量评估
-
FastQC软件:可以对fastq格式的原始数据进行质量统计。
-
官方网站—帮助文档
-
常用参数:
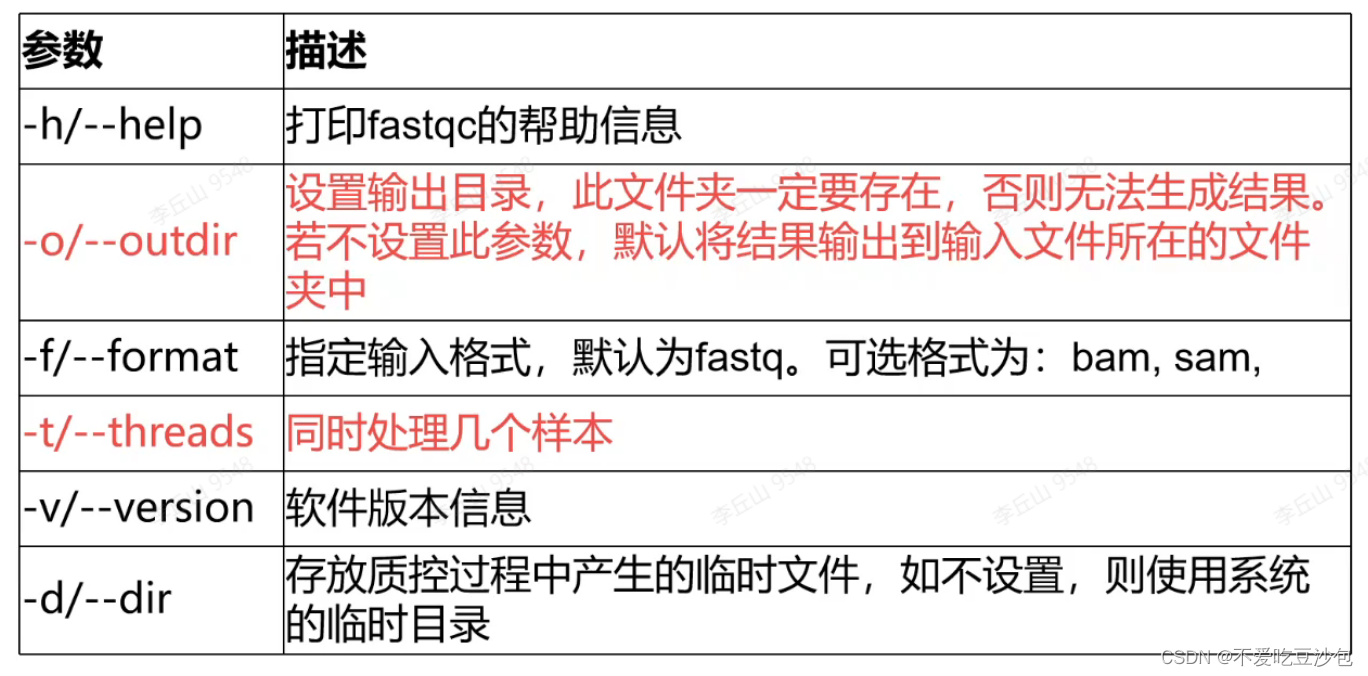
-
任务运行
- 在当前窗口直接运行
- 将命令后台运行
nohup ... & - 将命令写入
sh脚本,使用nohup ... &运行脚本
-
绝对路径运行脚本
multiqc=/home/t_rna/miniconda3/envs/rna/bin/multiqc fastqc=/home/t_rna/miniconda3/envs/rna/bin/fastqc fq_dir=$HOME/project/Human-16-Asthma-Trans/data/rawdata outdir=$HOME/project/Human-16-Asthma-Trans/data/rawdata # 使用绝对路径运行 $fastqc -t 6 -o $outdir ${fq_dir}/SRR*.fastq.gz >${fq_dir}/qc.log -
确定后台任务结束
- 看结果目录有无正常结果生成
htop中是否有命令正在运行ps fx看本人的进程- 读输出的运行日志
-
质控后生成的html文件即质控报告
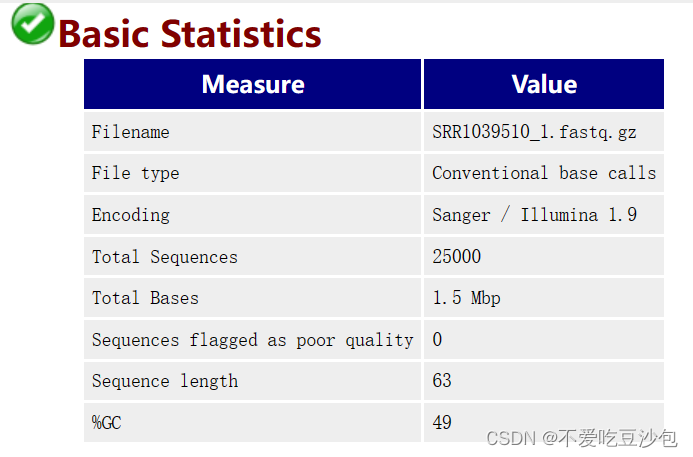
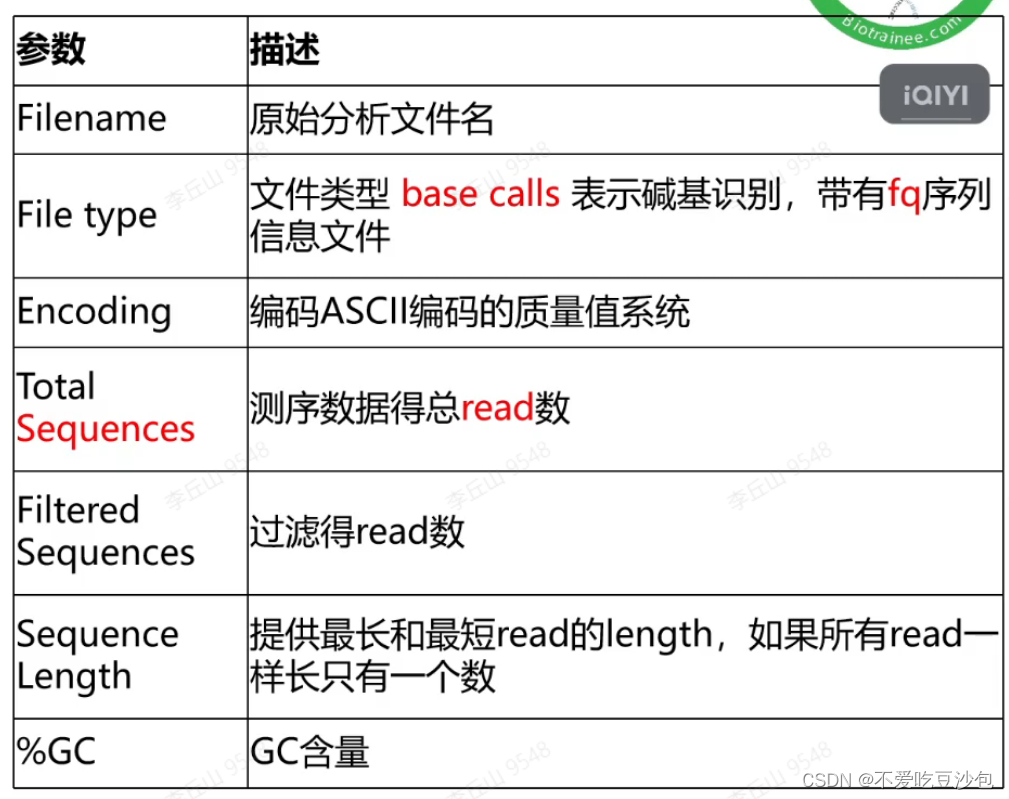
数据量统计方式:生物学中碱基数量VS计算机中的存储字节(需要加以区分)
-
per base sequence quality
按位置展示碱基质量的箱线图
-
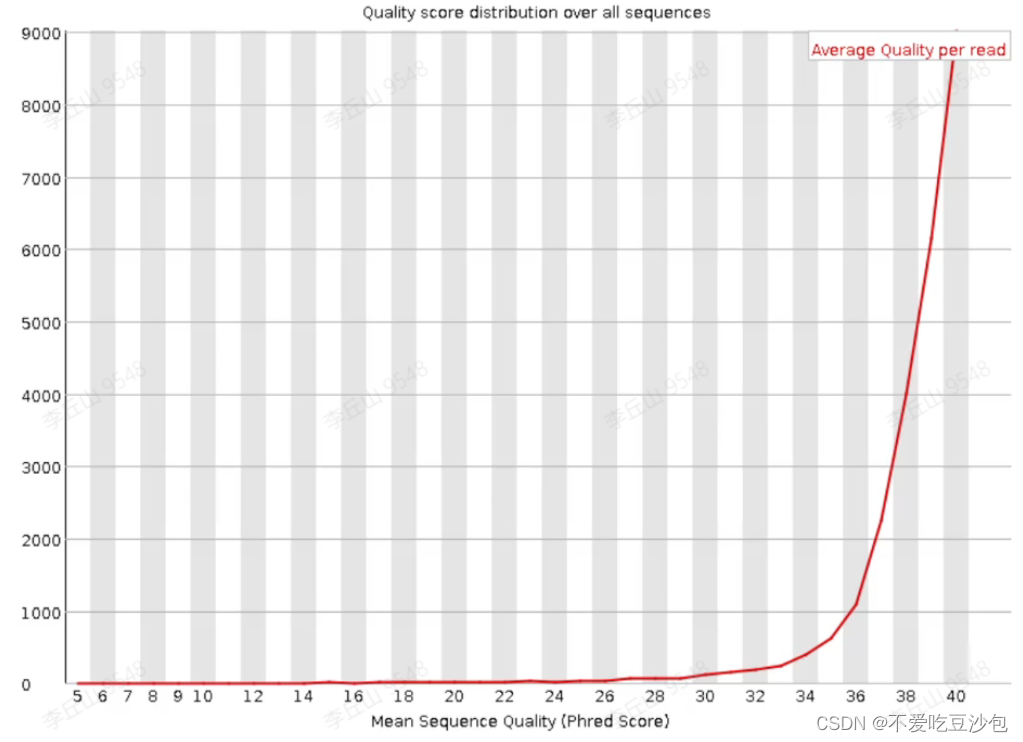
per sequence quality scores
横坐标:一条序列碱基的平均Q值(一条序列中N越多,质量值越低)
纵坐标:每个质量值对应的read数
较好结果:
-
per base sequence content
每个碱基位置上:ATGC含量的分布图
理论上G/C、A/T的含量在测序循环上应分别相等,且稳定呈水平线。
差数据:出现分离。
-
per sequence GC content
GC含量分布图,蓝色和红色线分别为理论值和实际值,越接近越好;一般呈单峰分布。
-
Atapt
公司提供数据
-
-
整合FastQC结果
multiQC *zip
-
数据过滤
原始序列质量控制标准:
- 去除含接头的reads;
- 过滤去除低质量值数据,确保数据质量;
- 去除含有N(无法确定碱基信息)的比例大于5%(可以自定义)的reads。
过滤接头
-
trim_galore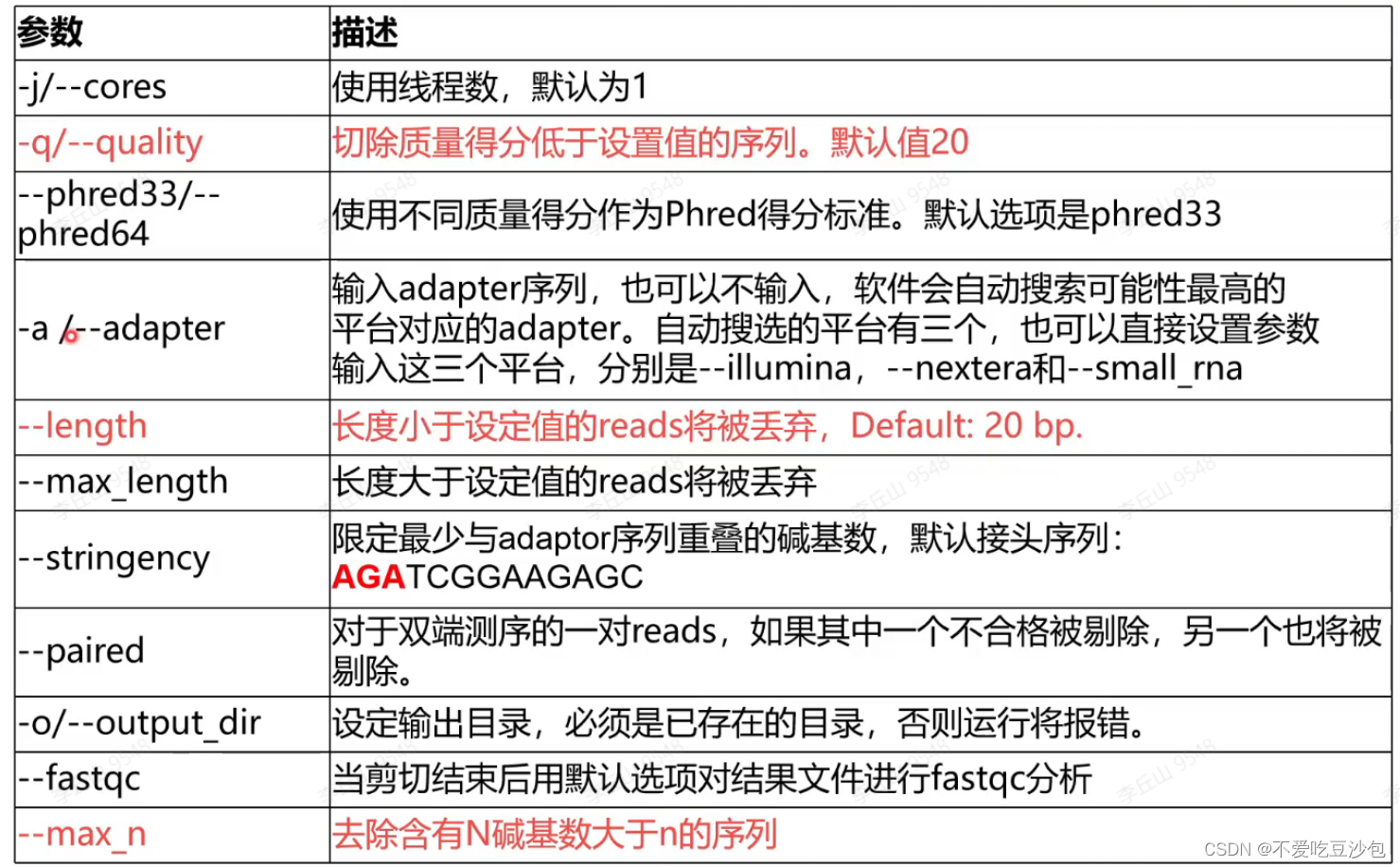
-
单个样本的运行
trim_galore -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ./ ../../rawdata/SRR1039510_1.fastq.gz ../../rawdata/SRR1039510_2.fastq.gz -
多个样本的运行
-
先获得样本名组成的文件
ID -
while循环
# 多个样本 vim trim_galore.sh,以下为sh的内容 rawdata=$HOME/project/Human-16-Asthma-Trans/data/rawdata cleandata=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore cat ID | while read id do trim_galore -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ${cleandata} ${rawdata}/${id}_1.fastq.gz ${rawdata}/${id}_2.fastq.gz done
-
-
-
Tips:
后台任务转前台:
jobs列出任务;fg %1(任务序号)前台任务转后台:
Ctrl+Z暂停,jobs列出任务,bg %1(任务序号)















 5496
5496











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









