






写在前面
我们还是在正式进行代码操作前想几个小问题:👇
- 如何将单细胞数据导入
R中?- 不同类型的数据/信息(如细胞信息、基因信息等)是如何存储和操作的?
- 如何获得细胞和基因的基本信息并对数据进行相应的过滤?
用到的包
目前常用的scRNA-seq分析包,包括Seurat、Scanpy(python)、Scater、Monocle2、Monocle3等。🤒
rm(list = ls())
library(tidyverse)
library(SingleCellExperiment)
library(DropletUtils)
数据格式
scRNA-seq的标准格式为SingleCellExperiment。 😘
Note! Seurat包有其自己的格式,即Seurat格式,可能因为Seurat太火了吧,越来越多的包都开始兼容Seurat格式的文件了。😂
我们拿到的数据通常是一个feature-by-sample的表达矩阵。
在scRNA-seq分析中,我们一般需要从counts矩阵开始分析,代表每个cell的feature的reads/UMI。feature可以是gene、isoforms或exons。🤒

SingleCellExperiment对象
4.1 读入数据
counts <- read.table("./molecules.txt", sep = "\t")
annotation <- read.table("./annotation.txt", sep = "\t", header = TRUE)
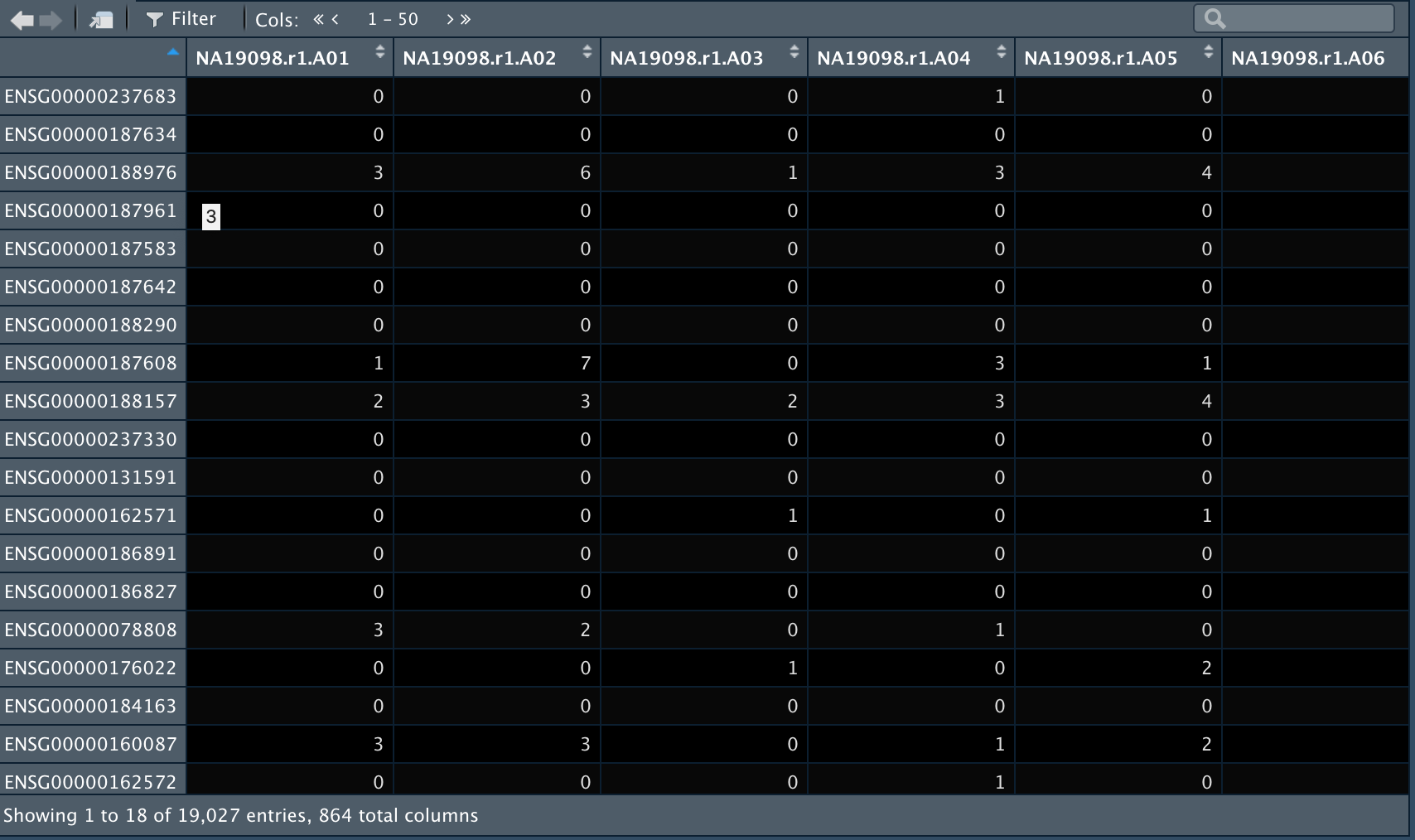

4.2 创建SingleCellExperiment对象
# 注意assays必须是matrix
tung <- SingleCellExperiment(
assays = list(counts = as.matrix(counts)),
colData = annotation
)
tung

Note! 这个SingleCellExperiment包含:
- 19027个基因(行)和864个细胞(列);
- 一个名为
counts的assays; - 预览部分基因名(
rownames)和细胞名(colnames); - 基因的
metadata为空 (rowData); - 细胞的
metabdata(colData names)
10X 文件的读取
像上面那样创建SingleCellExperiment对象,几乎适用于任何情况,我们只需要一个counts矩阵即可。 🤗
但有时候我们获取的文件是cellranger(用于10X Chromium数据)的输出文件,这个时候我们可以用DropletUtils包中的read10xCounts函数。😀
sce <- read10xCounts("./pbmc_1k_raw")
sce <- read10xCounts("./pbmc_1k_filtered")
sce

查看数据
6.1 查看counts矩阵
这里需要说一下的是,如果你的矩阵就叫counts,那么counts(tung) = assay(tung, "counts")。
counts(tung)
assay(tung, "counts")[1:4,1:4]

6.2 查看cell信息
colData(tung)

6.3 查看batch信息
table(tung$batch)
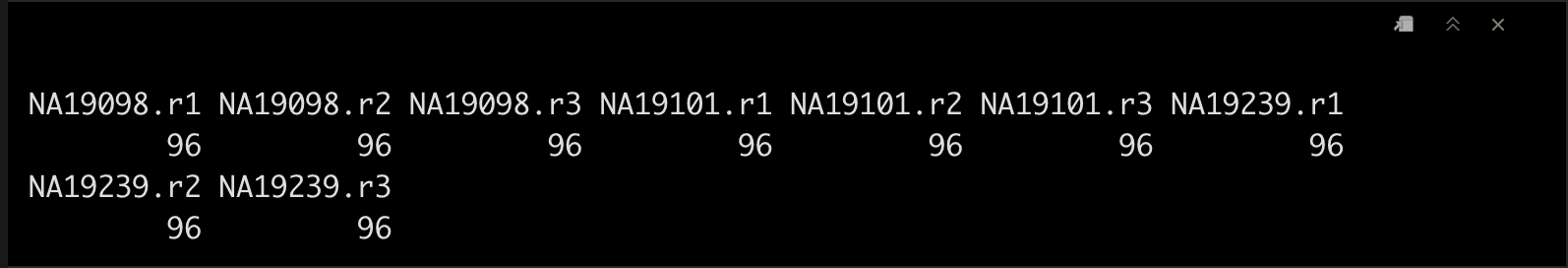
🧐 补充!
| Function | Description |
|---|---|
rowData(sce) | gene信息 |
colData(sce) | cell信息 |
assay(sce, "counts") | 查看counts矩阵 |
reducedDim(sce, "PCA") | 查看PCA降维table |
sce$colname | 查看colData的colname, 等价于colData(sce)$colname |
sce[<rows>, <columns>] | 查看特定行、列 |
修改SingleCellExperiment对象
举个栗子 🌰
我们把counts矩阵进行log2(x+1)转换,命名为logcounts。
assay(tung, "logcounts") <- log2(counts(tung) + 1)
# 查看一下
logcounts(tung)[1:4, 1:4]

🧐 补充!
| Code | Description |
|---|---|
assay(sce, "name") <- matrix | 添加新矩阵 |
rowData(sce) <- data_frame | 替换rowData |
colData(sce) <- data_frame | 替换colData |
colData(sce)$column_name <- values | 在colData中添加/替换一个新的列 |
rowData(sce)$column_name <- values | 在rowData中添加/替换一个新的行 |
reducedDim(sce, "name") <- matrix | 添加降维矩阵 |
基本数据处理
8.1 举个栗子🌰
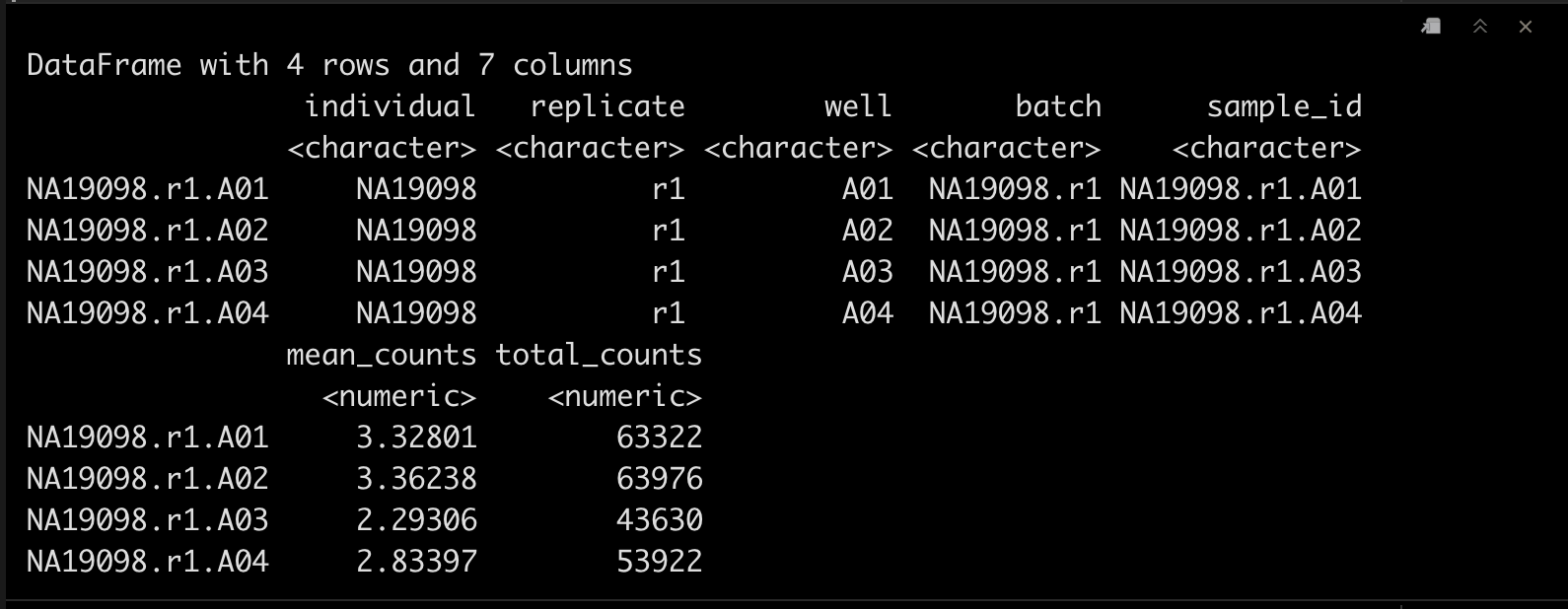
计算我们的数据集中每个细胞(列)的平均counts。
然后我们将它加到column metadata中作为新的一列。😘
colData(tung)$mean_counts <- colMeans(counts(tung))
这个时候我们可以查看一下,多了一列mean_counts。🤔
colData(tung)[1:4,]
8.2 补充一下
这里我们再补充一下基本函数。有兴趣的小伙伴都试试吧。🫶
- row (feature) summaries
rowSums(counts(tung)) # sum
rowMeans(counts(tung)) # mean
rowSds(counts(tung)) # standard deviation
rowVars(counts(tung)) # variance
rowIQRs(counts(tung)) # inter-quartile range
rowMads(counts(tung)) # mean absolute deviation
- column (sample) summaries
colSums(counts(tung)) # sum
colMeans(counts(tung)) # mean
colSds(counts(tung)) # standard deviation
colVars(counts(tung)) # variance
colIQRs(counts(tung)) # inter-quartile range
colMads(counts(tung)) # mean absolute deviation
CPM的计算与查看
9.1 CPM的计算
这里我们直接使用sweep函数来搞定counts转CPM,在这之前我们先计算一下total counts并新增一列。
colData(tung)$total_counts <- colSums(counts(tung))
assay(tung, "cpm") <- sweep(counts(tung),2,tung$total_counts/1e6,'/')
# check一下
colSums(cpm(tung))[1:10]

9.2 CPM矩阵的查看
cpm(tung)[1:4, 1:4]
assay(tung, "cpm")[1:4, 1:4]

常用过滤方法
拿到矩阵后,我们一般需要过滤一些低表达或异常表达值。
举个栗子 🌰,假设细胞至少25000counts,基因在至少一半的细胞中拥有超过5个counts。
10.1 过滤细胞
cell_filter <- colSums(counts(tung)) >= 25000
# check一下
table(cell_filter)

10.2 过滤基因
gene_filter <- rowSums(counts(tung) > 5) > ncol(tung)/2
# check一下
table(gene_filter)

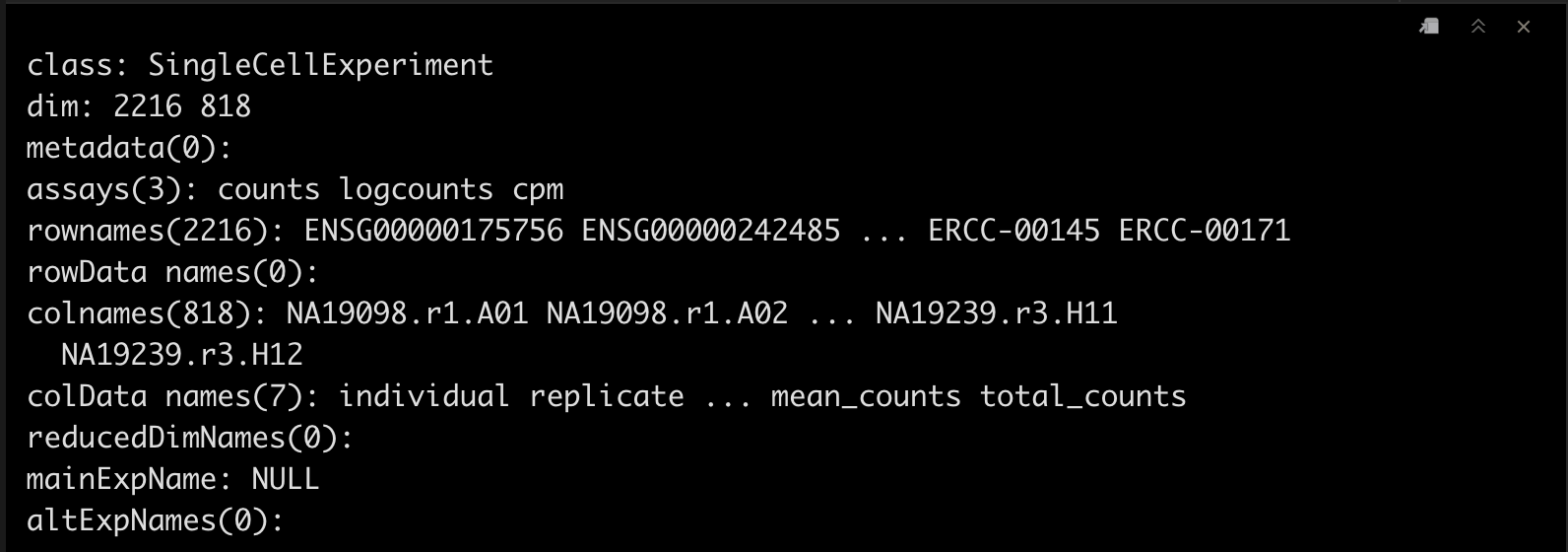
10.3 最终过滤
tung_filtered <- tung[gene_filter, cell_filter]
tung_filtered
10.4 补充一下
colSums(counts(sce)) > x # 对细胞进行过滤, Total counts per cell greater than x。
colSums(counts(sce) > x) > y # 对细胞进行过滤,Cells with at least y genes having counts greater than x.
rowSums(counts(sce)) > x # 对基因进行滤过,Total counts per gene greater than x.
rowSums(counts(sce) > x) > y # 对基因进行过滤,Genes with at least y cells having counts greater than x.

需要示例数据的小伙伴,在公众号回复
scRNAseq获取吧!点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
本文由mdnice多平台发布















 2230
2230

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









