






单细胞数据处理记录
size_factor计算
这个计算式来自scanpy包的,方式,是计算细胞的。
if size_factors:
sc.pp.normalize_per_cell(adata)
adata.obs['size_factors'] = adata.obs.n_counts / np.median(adata.obs.n_counts)
这个计算是来自2023年nature methods论文:
Comparison of transformations for
single-cell RNA-seq data
他的公式:
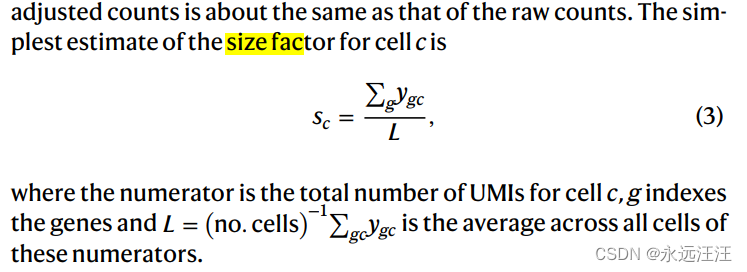
经过我的理解,编写的代码:
import numpy as np
#cell*gene [2*3]
matrix = np.array([[1, 2, 3], [4, 5, 6]])
# 其中分子是细胞c的 UMI 总数,g索引基因和L是这些分子的所有细胞的平均值。
def size_factor(x):
gene_sum=x.sum(1)# [6, 15]where the numerator is the total number of UMIs for cell c,
cell_L=x.sum()/matrix.shape[0]
sf=gene_sum/cell_L
return gene_sum,x.sum(),cell_L,sf
size_factor(matrix)
运行结果:(array([ 6, 15]), 21, 10.5, array([0.57142857, 1.42857143]))
但是我的数据,是经过log1p变化的,或者其他log(x+1)变化的。可以通过np.expm1()逆变换过来,举例如下。
np.log1p(matrix+1),np.expm1(np.log1p(matrix+1))-1
结果:
(array([[1.09861229, 1.38629436, 1.60943791],
[1.79175947, 1.94591015, 2.07944154]]),
array([[1., 2., 3.],
[4., 5., 6.]]))
规范化计算
参考那篇nature文章的规范化方法,目前是相对最好的规范化方法。文章的核心不是普通的规范化log(x+1),而是log(x/s +1);

编写代码如下:
import numpy as np
matrix = np.array([[1, 2, 3],
[4, 5, 6]])
sf = size_factor(matrix) # 计算每个细胞的size_factor
normalized_matrix = np.log1p(matrix / sf[:, np.newaxis]+1) # 每个元素除以对应的size_factor
print(sf)
print(normalized_matrix)
[0.57142857 1.42857143]
[[1.32175584 1.70474809 1.98100147]
[1.56861592 1.70474809 1.82454929]]















 9368
9368











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









