






runHiC是python包,用来分析HiC数据。
HiC(High-throughput chromosome conformation capture)是高通量染色体构象捕获技术的英文简称。HiC数据可以将scaffolds锚定到染色体上,即将基因组组装到染色体水平。
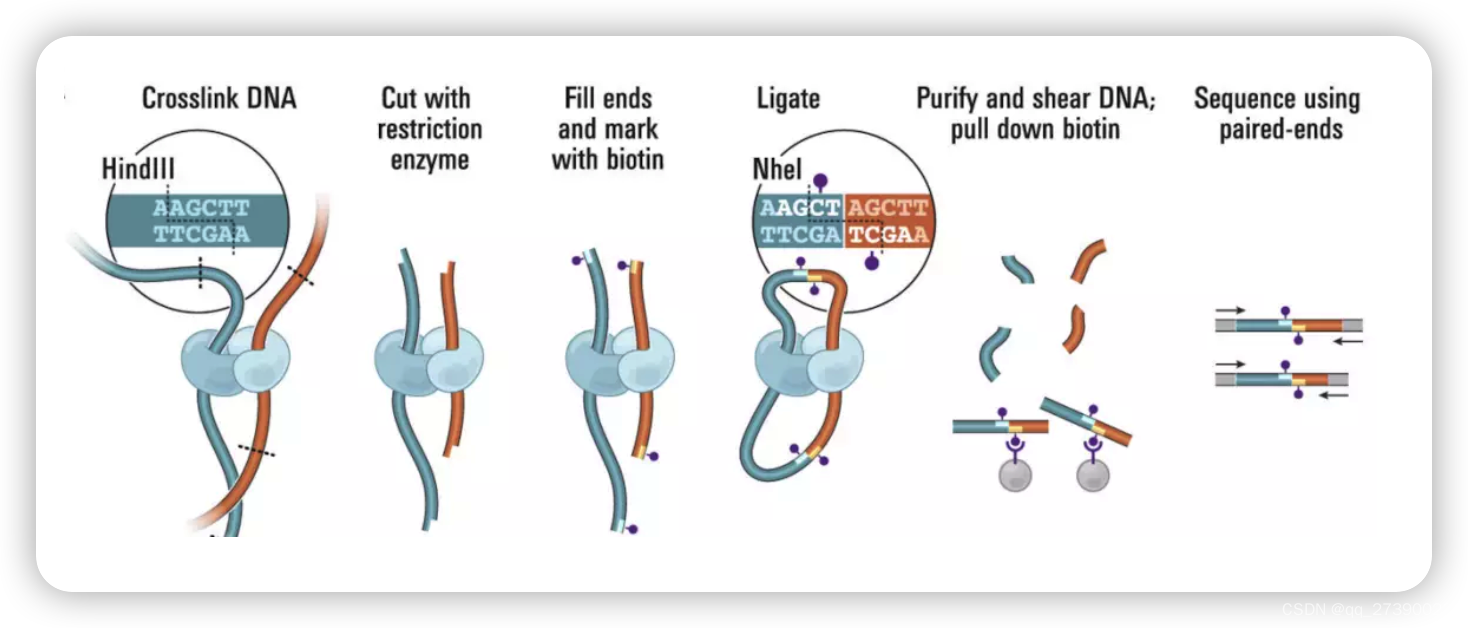
1. 安装runHiC环境
conda create --name HiC_seq python=3.8
# 安装包
# conda install
# numpy=1.20 pandas cooler=0.8.6 matplotlib biopython pairtools bwa sra-tools
# minimap2 samtools pigz chromap
# Conda 安装不上的用pip
pip install runHiC
pip install cooler=0.8.6
#pip install pip-search
#pip_search pairtools
2. 下载测试数据,基因组数据
mkdir HiC_workspace
mkdir HiC_data
cd HiC_data
mkdir test_fq_data
cd test_fq_data
prefetch SRR027958 &
prefetch SRR027956 &
fastq-dump --split-files SRR027958/SRR027958.sra
for i in SRR027958/*.fastq; do gzip -c $i > SRR027958/`basename $i`.gz; done
fastq-dump --split-files SRR027956/SRR027956.sra
for i in SRR027956/*.fastq; do gzip -c $i > SRR027956/`basename $i`.gz; done
# 下载参考基因组数据
Cd ..
mkdir hg38
cd hg38
wget ftp://hgdownload.cse.ucsc.edu/goldenPath/hg38/chromosomes/*
###Python代码##########
>>> import os, glob
>>> labels = list(map(str,range(1,23))) + ['X','Y','M']
>>> pool = ['chr{0}.fa.gz'.format(i) for i in labels]
>>> for c in glob.glob('*.fa.gz'):
... if not c in pool:
... os.remove(c)
>>> exit()
###Python代码 end #######
gunzip *.gz
cat *.fa > hg38.fa
# 可以删除每一个染色体的序列文件3. mapping
- reads对将用bwa-mem映射到hg38参考基因组,比对结果将以BAM格式报告,并置于比对-hg38下。
- BAM文件将使用pairs-hg38下的pairtools解析为.pairs。
在对齐分析过程中,runHiC检测结扎连接,标记各种情况(未映射、多映射、多结扎行走和有效的单结扎),并对对进行排序以进行进一步分析。在本例中,.pairsam。pairs-hg38下的gz文件是4DN联合体提出的有效.pairs文件。默认情况下,它只包含7列:chr1、pos1、chr2、pos2、strand1、strand2和pair_type;如果在命令中添加--include readid,您将得到一个额外的“readid”列;如果指定--include sam,将添加两个额外的列“sam1”和“sam2”来存储原始对齐;如果添加--drop seq,seq和QUAL将从sam字段中删除,以节省磁盘空间。
cd ../../HiC_workspace
mkdir HiC_workspace
runHiC mapping -p ../HiC_data -g hg38 -f test_fq_data -F FASTQ -A bwa-mem -t 32 --include-readid --logFile runHiC-mapping.log 参数:
-p DATAFOLDER, --dataFolder DATAFOLDER
Path to the root data folder. Both sequencing reads and reference
genome should be placed under this folder. (default: None)
-g GENOMENAME, --genomeName GENOMENAME
Name of the folder containing the reference genome fasta file.
(default: None)
如hg38/hg38.fa
-f FASTQDIR, --fastqDir FASTQDIR
Name of the folder containing sequencing reads. (default: None)
-A {bwa-mem,chromap,minimap2}, --aligner {bwa-mem,chromap,minimap2}
Name of the sequence alignment software to invoke. (default:
chromap)
-t THREADS, --threads THREADS
Number of threads. (default: 8)
--include-readid If specified, add read IDs to the outputed .pairsam files.
(default: False)
--chunkSize CHUNKSIZE
On a low-memory machine, it's better to split the raw read file
into chunks and map them separatively. This parameter specifies
4.Filtering
runHiC的筛选子命令旨在对对齐的读取对执行基本筛选过程:
- 去除多余的PCR伪影。
- 删除映射到同一限制片段的读取对。
runHiC filtering --pairFolder pairs-hg38/ --logFile runHiC-filtering.log --nproc 32
5.binning
在此阶段,将在coolers-hg38子文件夹下为每个.pairs生成一个.mcool文件。gz文件使用cooler器。mcool格式是4DN联盟的官方Hi-C数据格式,可以使用HiGlass可视化。
runHiC binning -f filtered-hg38/ --logFile runHiC-binning.log --nproc 326. 一步完成(mapping + filtering + binning)
runHiC pileup -p ../HiC_data -g hg38 -f test_fq_data -F FASTQ -A bwa-mem -t 32 --include-readid --logFilerunHiC.log参考:















 1080
1080











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









