










 本文引入新的谱聚类框架用于单细胞RNA测序(scRNA-seq)数据分析。该方法在目标矩阵施加稀疏结构,利用多个双随机相似矩阵学习相似矩阵,通过ADMM算法迭代求解非凸问题。在模拟和真实scRNA-seq数据上评估,结果表明该方法能准确、稳健地识别聚类。
本文引入新的谱聚类框架用于单细胞RNA测序(scRNA-seq)数据分析。该方法在目标矩阵施加稀疏结构,利用多个双随机相似矩阵学习相似矩阵,通过ADMM算法迭代求解非凸问题。在模拟和真实scRNA-seq数据上评估,结果表明该方法能准确、稳健地识别聚类。
Abstract
Motivation:单细胞rna测序(scRNA-seq)技术可以在单细胞水平上生成全基因组表达数据。scRNA-seq分析的一个重要目标是对细胞进行聚类,其中每个聚类由基于基因表达模式的属于相同细胞类型的细胞组成。
Results:我们引入了一种新的谱聚类框架,它在目标矩阵上施加稀疏结构。具体来说,我们利用多个双重随机相似矩阵来学习相似矩阵,其动机是观察到每个相似矩阵可以是数据的不同信息表示。我们在目标矩阵上施加一个稀疏结构,然后缩小目标矩阵中行对差(pairwise differences of the rows),这是因为目标矩阵在理想情况下应该具有这些结构。利用ADMM算法迭代求解该非凸问题,并证明了算法的收敛性。我们在各种模拟和真实的scRNA-seq数据上评估了所提出的聚类方法的性能,并表明它可以准确和鲁棒地识别聚类。
1 Introduction
单细胞测量的最新进展帮助科学家更好地了解细胞异质性(如Kalisky和Quake,2011;Pelkmans, 2012)。然而,单细胞数据集带来了统计和计算方面的挑战,例如使用单细胞数据来识别基因表达谱中反映的相同功能状态的细胞组。从单细胞数据中识别子群是一个无监督分类问题,主成分分析(PCA)、光谱聚类(von Luxburg, 2007)和k-means (Forgy, 1965)是最常用的子群识别方法。然而,与大量RNA-seq或基因表达微阵列相比,单细胞RNA-seq (scRNA-seq)数据的一个主要挑战是,由于技术和采样问题,它们具有高水平的噪声和许多缺失值。即使在相同类型的细胞中,基因表达水平的高度可变性也会混淆这些现有的聚类方法。
为了解决scRNA-seq数据分析中的这些问题,已经提出了几种新的聚类方法。例如,Xu和Su(2015)提出了一种具有共享近邻相似性的基于派系的方法,该方法在识别细胞类型方面表现出更高的性能。已经提出了涉及迭代聚类的复杂方法来进行亚型分类和检测亚型之间的关系。Haghverdi等人(2015)通过扩散图对数据进行了降维,扩散图强调细胞状态沿着假定的发育途径的连续性。Shao和Ho¨fer(2017)利用非负矩阵分解(NMF)技术将高维单细胞数据分解为生物可解释的成分。NMF检测功能细胞亚群,同时指导识别数据中的生物学相关特征。Wang等人(2017)通过多核学习(SIMLR)提出了单细胞解释,该方法从scRNA-seq数据中学习相似性度量,以执行降维和聚类。SIMLR的关键特征是学习相似矩阵并利用多核分离聚类。这些积极的方法发展反映了从scRNA-seq数据中无监督学习生物学相关特征的许多挑战。
谱聚类(SC)是一种流行的现代聚类方法,它利用从数据中导出的矩阵的特征向量进行聚类。SC易于实现,可以通过标准线性代数软件有效地求解,并且通常优于传统的聚类算法,如k-means算法。尽管有这些优势,SC的结果对相似性度量的选择很敏感,并且从scRNA-seq数据中获得合适的相似性度量需要额外的努力。
现有的提高SC性能的方法可分为两种方法: (i)在数据相似度矩阵固定的情况下提高SC聚类精度;(ii)构建合适的相似度矩阵来提高聚类性能。
在本文中,我们提出了一种在这两方面都进行了改进的新方法。
关于第一种方法,我们通过在目标矩阵上施加稀疏结构来修改SC框架。这是由于观察到,这种结构对于更好的聚类性能至关重要,但当数据包含高水平的噪声时,SC通常无法获得(Lu et al.,2016a;Wang et al.,2017)。
关于第二种方法,我们利用多个双随机亲和矩阵来构造一个鲁棒相似度矩阵。这可以帮助获得更准确和稳健的聚类结果,即使数据包含许多缺失的值和不平衡的相似性,通过标准化的相似性矩阵,使所有数据点的总相似性相等(例如图1)。
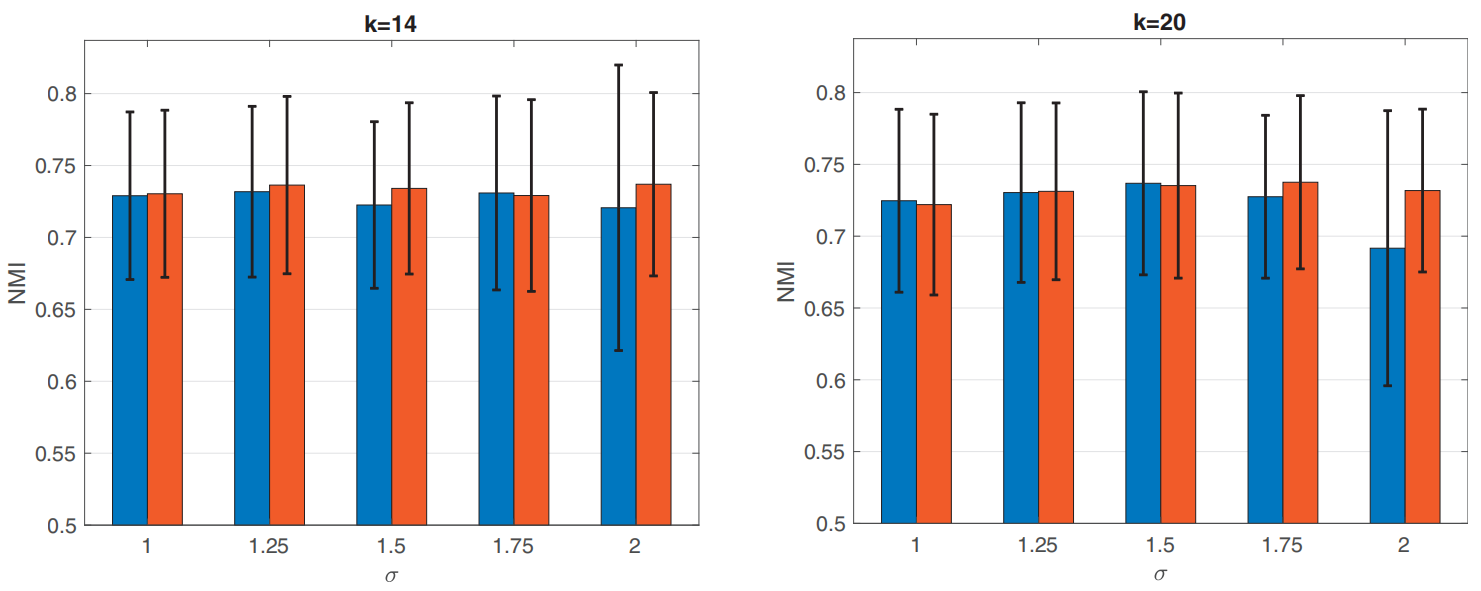
图1: sigma是相似矩阵的核参数,蓝色条形图是标准的归一化相似度矩阵,橘色条形图是双随机相似度矩阵,数据缺失比例为78.6%
2 Materials and methods
2.1 Spectral clustering
给定一组数据点X其中n是样本数量,p是数据的维度,光谱聚类(SC)使用相似矩阵
S
=
s
i
j
S=sij
S=sij, 其中
s
i
j
sij
sij表示数据点xi和xj之间的相似性度量。为了使SC表现良好,重要的是选择一个合适的相似矩阵
S
S
S。高斯核函数是构造
S
S
S最广泛使用的函数之一。为了将数据X划分为C个簇,SC解决了以下优化问题:
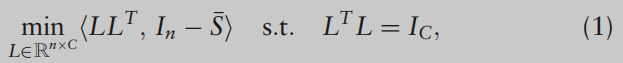
注意,在理想情况下,标准正交矩阵L,即(1)的解,应该具有稀疏结构,使得样本i属于第j个聚类时
L
i
j
≠
0
L_{ij} \ne 0
Lij=0。因此,LLT应该是一个块对角矩阵,因此具有稀疏结构。根据这一观察结果和
∥
L
L
T
∥
F
2
=
t
r
(
L
L
T
)
=
C
\left \| LL^T \right \| ^2_F=tr(LL^T)=C
LLT
F2=tr(LLT)=C 的事实,可以考虑以下(1)的正则化版本来找到更好的U:
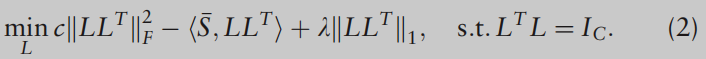
在这里,是否添加第一项在数学上是等价的,但是这个项为所提出的算法提供了更理想的收敛特性,这将在第2.3节中介绍。
因为(2)包含了一个非线性约束
L
T
L
=
I
C
L^TL=I_C
LTL=IC,所以它不是凸的。为了解决非凸模型的计算问题,我们遵循稀疏谱聚类的思想(Lu et al.,2016a),它为稀疏谱聚类添加了松弛的凸约束:
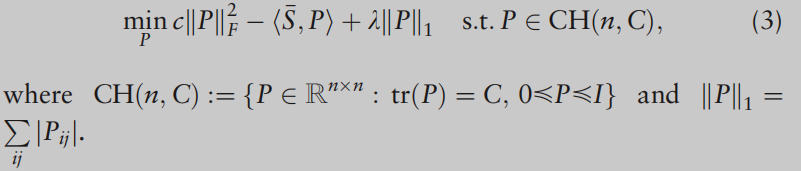
REMARK 1
Lu等人(2016a)也使用了这样一个事实,即集合CH(n, C)是集合
{
P
=
L
L
T
∈
R
n
×
n
:
L
T
L
=
I
C
}
\{P=LL^T \in R^{n \times n}: L^TL=I_C \}
{P=LLT∈Rn×n:LTL=IC}的凸包。值得注意的是,(3)由于附加项
∥
P
∥
F
2
\left \| P \right \|^2_F
∥P∥F2是严格凸的,因此使用多个相似矩阵的(3)的广义公式(非凸)可以通过具有收敛保证的迭代算法来计算。详细信息请参见2.3.1节。
2.2 Multiple similarity learning
由于单细胞数据的复杂性,依赖于单个相似性可能不能提供足够的信息,我们可以从考虑多个相似性矩阵中获益。此外,SC的性能对数据点之间的单一相似性度量很敏感,并且没有明确的标准来选择最佳相似性度量。继Wang et al.(2017)之后,我们考虑多个核函数来构建相似矩阵,如下所示:对于样本i和j,
1
≤
i
≤
j
≤
n
1 \le i \le j \le n
1≤i≤j≤n,
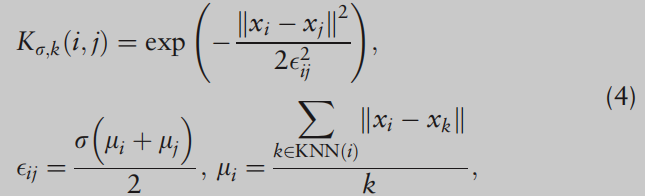
因此,使用多个核函数的广义框架比使用单个核函数更能适应所分析的数据。在本文中,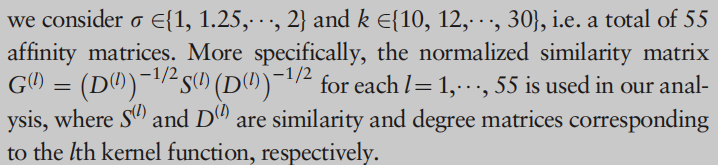
2.3 Proposed method
在本节中,我们提出了提出的方法的三个步骤。
步骤1:构造对称双随机相似矩阵
利用对称双随机亲和矩阵构造归一化图拉普拉斯矩阵。请注意,双随机相似矩阵已用于改进聚类分析(Zass和Shashua,2006;Lu et al., 2016b)。这种归一化的动机是由于流行的亲和矩阵归一化[例如normalization -cuts (Shi和Malik, 2000)]与双随机约束(Zass和Shashua, 2006)相关联。Lu et al. (2016b)表明,双随机相似矩阵输入的t-SNE (van der Maaten and Hinton, 2008)倾向于在嵌入空间中提供较少拥挤的样本。我们观察到,对于图拉普拉斯,使用双随机亲和矩阵的SC的性能与使用归一化图拉普拉斯相似或更好(图1)。我们将Sinkhorn-Knopp迭代算法(SK算法)(Sinkhorn和Knopp, 1967)应用于每个 l l l 的归一化亲和矩阵 G ( l ) G^{(l)} G(l),并获得对称的双随机矩阵 G ˉ ( l ) \bar{G}^{(l)} Gˉ(l)。如果SK算法具有输入矩阵 G ( l ) G^{(l)} G(l)的稀疏结构,则SK算法可以保持输入矩阵 G ( l ) G^{(l)} G(l)的稀疏结构。注意,SK算法生成一个矩阵序列,其行和列交替规范化。
第二步:进行稀疏谱聚类
在第二步中,我们考虑通过将对称双随机矩阵
G
ˉ
(
l
)
\bar{G}^{(l)}
Gˉ(l) 与相似性学习相结合来进行优化(3):

REMARK 2.
当
ρ
\rho
ρ 趋于无穷时,所有
w
l
w_l
wl 的权值相等。注意,
∑
l
w
l
G
ˉ
(
l
)
\sum_l w_l \bar{G}^{(l)}
∑lwlGˉ(l) 仍然是一个对称的双随机矩阵。人们可以对
w
l
w_l
wl 使用不同的正则化,但是使用惩罚
∑
l
w
l
G
ˉ
(
l
)
\sum_l w_l \bar{G}^{(l)}
∑lwlGˉ(l) 可以在迭代算法中得到
w
l
w_l
wl 的封闭解,从而减少了计算时间。
第三步3:缩小目标矩阵的两两差值
在这一步中,我们利用(1)中的理想
L
L
T
LL^T
LLT 是一个块对角矩阵,因此有许多相等的行向量。我们考虑以下优化:对于某个惩罚参数
μ
>
0
\mu > 0
μ>0,
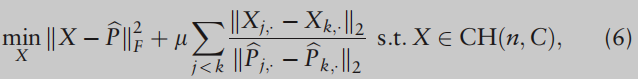
其中(6)中的成对融合惩罚自适应地将
∥
X
j
,
⋅
−
X
k
,
⋅
∥
\left \|X_{j,\cdot} - X_{k,\cdot}\right \|
∥Xj,⋅−Xk,⋅∥ 中的部分自适应地缩小为零,这是自适应Lasso的基本思想(Zou, 2006)。设
X
^
∈
R
n
×
n
\hat{X} \in R^{n \times n}
X^∈Rn×n 为(6)的解。我们通过取前C个特征向量对应于
X
^
\hat{X}
X^ 的前C个最大特征值得到
L
^
∈
R
n
×
C
\hat{L} \in R^{n \times C}
L^∈Rn×C,并对
L
^
\hat{L}
L^ 的归一化规范应用k-means来找到n个样本的隶属性。注意(6)也是凸的,可以使用ADMM计算。。
请注意,Wang等人(2017)对目标相似矩阵施加了低秩约束以获得块对角线结构。但是,低秩矩阵并不一定具有块对角结构。我们施加了更强的约束来获得块对角结构,因为这种结构对于更好的聚类性能是必不可少的。本文提出的光谱聚类与Lu et al. (2016a)的不同之处在于以下三个方面:第一步,将归一化亲和矩阵转化为对称双随机矩阵;在第二步中,我们使用自适应Lasso型惩罚项,并加入额外的二次项 ∥ P ∥ F 2 \left \| P \right \|^2_F ∥P∥F2;在第三步中,我们的目标是使用成对融合惩罚来获得一个行方向相似的目标矩阵,以获得块对角结构。
REMARK 3.
可以考虑以下两步优化,而不是两步优化
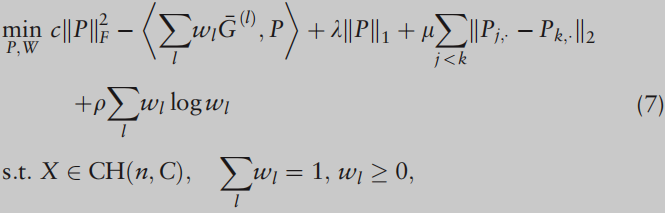
它也是凸的,可以使用ADMM算法进行计算。但所提出的两步法更有利,因为它在第三阶段使用输出矩阵
P
^
\hat{P}
P^,自适应地惩罚行差的欧氏范数。
2.3.1 Algorithm

虽然(5)不是联合凸,但所提出的迭代算法(8)-(9)具有收敛性:
PROPOSITION 1.
设G(P, W)为(5)的目标函数,则迭代(Pi, Wi)收敛于G的全局极小点,其中目标值G(Pi, Wi)单调递减。
所提出的迭代算法的收敛性是由于给定P和W中的一个是固定的,G(P, W)具有唯一的全局最小值(补充材料的B节)。
2.4 Choosing the number of clusters
提出的聚类方法需要一个目标数量的聚类。我们使用以下过程来选择C。首先,我们使用足够大的数 C’ 作为目标数,通过求解(5)得到 P ^ \hat{P} P^。设 λ 1 ≥ λ 2 ≥ λ n ≥ 0 \lambda_1 \ge \lambda_2 \ge \lambda_n \ge 0 λ1≥λ2≥λn≥0 为 P ^ \hat{P} P^的特征值,定义 C = a r g m a x j { λ j − λ j + 1 } C=argmax_j \{ \lambda_j - \lambda_{j+1}\} C=argmaxj{λj−λj+1}。我们用C作为目标数,即寻找一个特征值差 P ^ \hat{P} P^ 较大的索引。使用单细胞数据集的经验证据表明,这种方法效果很好。
3 Simulation results
在本节中,我们提出仿真研究来评估所提出方法的性能。我们使用以下三个性能指标来评估获得的聚类与真实标签之间的一致性:归一化互信息(NMI) (Strehl和Ghosh, 2003),纯度和调整的兰德指数(ARI) (Wagner和Wagner, 2007b)。NMI和Purity的值在0到1之间,但是ARI可以产生负值。这些度量度量两个聚类标签的一致性,因此值越高表示一致性越高。有关这些指标的详细信息,请参见
补充材料D部分。
REMARK 4.
ARI是一种基于对象对计数的度量。注意,ARI依赖于对聚类分布的强假设;它假设一个广义的超几何分布作为零假设,即两个聚类随机绘制,每个聚类中有固定数量的聚类和固定数量的元素(Wagner and Wagner, 2007a)。纯度是依赖于映射的度量之一,它不是一对一的。这种映射可能偏向于具有最大规模的集群(Wagner and Wagner, 2007a)。NMI基于互信息,互信息起源于信息论,并以熵的概念为基础。请注意,NMI不存在基于计数对或映射的度量的缺点(Wagner and Wagner, 2007a)。
在实验中,我们使用了两种类型的模拟数据。我们使用以下四个步骤生成第一个仿真模型。在第一步中,我们在二维潜在空间中生成C个点来创建一个圆,每个点被认为是一个簇的中心。n个点是通过在相应的聚类中心加入独立的噪声而生成的。在第二步中,我们将生成的2维数据投影到表示基因表达数据的p维空间。在第三步中,我们通过添加独立高斯噪声来模拟一个有噪声的基因表达矩阵。在最后一步中,我们引入了一个dropout事件,使得每个条目都以一定的概率独立地被观察到。在第二个仿真模型中,我们使用高斯混合模型生成数据。为了区分不同的细胞类型,很可能只有部分基因是信息性的,而非信息性和高噪声的基因会增加识别细胞类型的难度。在此背景下,我们使用一些属性来区分仿真模型中的聚类标签。有关这些仿真模型的详细信息,请参见补充材料的E部分。
在第一个实验中,我们用正则归一化亲和矩阵和双随机矩阵得到的不同图拉普拉斯算子比较了SC的性能。图1显示了基于 k ∈ { 14 , 20 } k \in\{14,20\} k∈{14,20}和 σ ∈ { 1 , 1.25 , 1.5 , 1.75 , 2 } \sigma \in \{1,1.25, 1.5, 1.75, 2\} σ∈{1,1.25,1.5,1.75,2} 的十个选择亲和矩阵的SC的平均NMI值和一个标准差对于两个不同的图拉普拉斯算子。我们可以看到,基于双随机亲和矩阵的SC的性能与具有正则归一化亲和矩阵的SC相似或更好。此外,我们看到用于构建亲和矩阵的不同内核的聚类性能有所不同。这说明需要一种新的不依赖于单一相似性度量的谱聚类方法,为此,我们利用2.2节中介绍的多个相似性矩阵。
在第二个实验中,我们研究了所提出的聚类方法在正则化参数 c , k , λ , μ c,k,\lambda,\mu c,k,λ,μ 变化方面的聚类性能的鲁棒性;我们从{0.01,0.05,0.1,1}中选择 c c c,从{5,10,…, 80}中选择k, λ , μ \lambda, \mu λ,μ 来自 { 1 0 − 5 , 1 0 − 4 , 1 0 − 3 , 1 0 − 2 } \{10^{-5},10 ^{- 4},10 ^{- 3},10 ^{- 2}\} {10−5,10−4,10−3,10−2} 。我们看到,对于这些参数的变化,聚类结果是鲁棒的,并且通过多种不同的 c , k , λ , μ c,k,\lambda,\mu c,k,λ,μ 组合可以获得稳定的聚类结果(补充图S2)。具体来说,我们观察到当 c = 0.1 , k = 10 , λ = μ = 1 0 − 4 c=0.1,k=10,\lambda=\mu=10 ^{- 4} c=0.1,k=10,λ=μ=10−4 时,性能始终保持良好。因此,在下面的应用程序中,我们将使用这些值。请注意,Lu等人(2016a)也在其修改的SC公式中纳入了 λ \lambda λ 的敏感性分析,并建议使用 λ = 1 0 − 4 \lambda=10 ^{- 4} λ=10−4。 ρ \rho ρ 选择的灵敏度见补充图S2F。在实现中,我们修复了 ρ = 0.2 \rho=0.2 ρ=0.2 ,它在各种设置下都表现良好。对真实scRNA-seq数据集的这些参数的敏感性分析(Supplementary fig S6-S14)也显示了所提出的聚类方法对于这些参数变化的鲁棒性。
在第三个实验中,为了研究使用多个亲和矩阵进行相似学习的效果,我们更直观地展示了图像的热图 ∣ V V T ∣ = ( ∣ V i , ⋅ V j , ⋅ T ∣ ) i , j \left | VV^T\right |=(\left | V_{i,\cdot}V_{j,\cdot}^T\right |)_{i,j} VVT =( Vi,⋅Vj,⋅T )i,j,其中 V ∈ R n × C V \in R^{n\times C} V∈Rn×C 具有标准正交列,由对应于 P ^ \hat{P} P^ 的前C个最大特征值的C个特征向量组成。其中 P ^ ∈ R n × n \hat{P}\in R^{n\times n} P^∈Rn×n 为任意SC方法得到的目标矩阵。在理想情况下, ∣ V V T ∣ \left | VV^T\right | VVT 应具有块对角结构。图2显示了 ∣ V V T ∣ \left | VV^T\right | VVT 的热图:图2A和图B考虑了使用不同相似性矩阵的标准SC。图2C考虑了没有步骤3的提议方法。图2D采用第2.3节提出的方法。对于图2A和B,我们在图2D中使用的建议的谱聚类中选择核权最小(0.0022)和核权最大(0.032)的核。有趣的是,与图2A相比,图2B中的结构更接近于块对角结构,这表明所提出的方法倾向于给予提供更清晰块对角结构的核更大的权重。我们观察到,与图2A、B和C相比,图2D具有更清晰的块对角结构,这表明相似性学习和步骤3有助于恢复真实结构。
在最后的实验中,我们将提出的无相似学习方法(“PSSC”)和提出的方法(“MPSSC”)与以下四种现有方法进行了比较:t-SNE;SIMLR;光谱聚类(SC);和稀疏谱聚类(’ SSC ') (Lu et al., 2016a)。对于PSSC,我们使用55个考虑核的平均相似矩阵。对于SSC,我们使用Lu等人(2016a)建议的正则化参数。图3A和B显示了模拟模型1具有一个标准差(误差条)的平均NMI值。当 γ = 0.01 \gamma=0.01 γ=0.01 (缺失率为37%)时,MPSSC、PSSC、SIMLR和tSNE具有相似的NMI值,且优于SC和SSC。配对样本t检验显示,SC、SSC和PSSC与MPSSC的均值差异显著 P-values < 0.001),但t-SNE和SIMLR与MPSSC的均值差异不显著( P-values 分别为0.08和0.16)。当 γ = 0.006 \gamma=0.006 γ=0.006 (缺失率为90%)时,MPSSC优于其他方法。MPSSC其他方法的p值均显著( P-values < 0.002)。仿真模型2的结果(图3C和D)表明,MPSSC、PSSC和SSC的值高于其他方法。对于©,配对样本t检验表明,SC、t-SNE和SIMLR与MPSSC的平均差异显著( P-values < 0.001),但SSC和PSSC的 P-values 分别为0.81和0.55。对于(D), MPSSC的SC、t-SNE和SIMLR的P值显著( P-values < 0.001),但SSC和PSSC的P值分别为0.07和0.44。综上所述,我们观察到MPSSC和PSSC与其他方法相比始终具有更高的值,这表明MPSSC和PSSC可以准确而稳健地识别中的聚类。
4 Applications to single-cell RNA sequence data
在本节中,我们将提出的聚类方法应用于单细胞RNA-Seq数据集,并与现有的聚类方法进行比较,以证明其聚类性能。我们收集了9个scRNA-seq数据集,代表了几种类型的动态过程,如细胞分化、细胞周期和对外部刺激的反应。每个scRNA-seq数据包含的细胞标记是先验已知的或在各自的研究中得到验证。表1总结了9个数据集的特征。有关9个scRNA-seq数据集的详细描述,请参见补充资料的F部分。
我们将PSSC和MPSSC与其他方法进行比较,使用第3节中的三个指标。对于PSSC和MPSSC,我们首先使用第2.4节中提出的方法估算集群数量。对于其他方法,我们使用真实的聚类数来获得聚类结果。图4总结了6个小规模单细胞数据集的NMI和计算时间。在许多情况下,MPSSC和PSSC具有更高的NMI值,这表明它们通常比竞争对手表现更好。这表明,与其他竞争对手相比,所提出的方法可以更好地揭示细胞间的相似性和差异性结构。它们的计算时间也与其他方法相当。图5总结了三个更大规模数据集的NMI和计算时间。对于更大规模的数据集,由于时间复杂性和内存问题,我们已经进行了MPSSC和PSSC,没有进行第三步。我们观察到,MPSSC和PSSC方法仍然具有更高的NMI值,而计算时间与SSC和SIMLR相当。纯度和ARI测量见补充图S4-S5。
在这9个数据集中,我们主要根据聚类结果对这两个数据集进行分析。表1中的第一个数据集称为Ginhoux数据集(Schlitzer et al., 2015),包含251个树突状细胞祖细胞在三种细胞状态之一的11834个基因的表达值:单核细胞和树突状细胞祖细胞(mdp)、普通树突状细胞祖细胞(cdp)和前树突状细胞(PreDCs)。DC祖细胞来源于骨髓中的造血干细胞,在成为完全发育的DC之前会经历多种细胞状态的转变(Schlitzer et al., 2015)。数据集包含59个mds、96个cdp和96个predc。尽管树突状细胞在脊椎动物适应性免疫系统的激活中起着重要作用,但在这一过程中涉及的一些机制仍存在争议。图6A使用MPSSC在二维空间中显示细胞。
为了可视化,我们利用MPSSC获得的 P ^ \hat{P} P^ 进行概率度量:我们将 P ^ \hat{P} P^ 转换为对称联合概率 Q = ( q i j ) i , j Q=(q_{ij})_{i,j} Q=(qij)i,j,使 q i j = P ^ i j = ∑ k , l P ^ k l q_{ij}=\hat{P}_{ij}=\sum_{k,l} \hat{P}_{kl} qij=P^ij=∑k,lP^kl,并应用t-SNE学习尽可能反映相似性qij的二维映射。我们观察到,同一类型的细胞很好地聚集在一起,而一些不同类型的细胞混合在一起,难以区分,这也是使用其他方法时发现的。请注意,嵌入的数据点大约位于球体周围。当输入相似矩阵是双随机时,这种现象经常被观察到,这可以解决拥挤问题。
第二个数据集(Deng et al., 2014),在表1中称为Deng数据集,包括从不同着床前阶段的小鼠胚胎中分离的单个细胞的转录组。数据包括135个细胞和12548个基因,细胞属于受精卵、2细胞早期、2细胞中期、2细胞晚期、4细胞期、8细胞期和16细胞期。如图6B所示,MPSSC组在2细胞早期、2细胞中期、2细胞晚期和4细胞阶段的合子都很好。然而,由于不同文库制备方案的技术差异,8细胞和16细胞阶段无法区分,Xu和Su(2015)也观察到这一点。这也可以解释为只有少数基因在8细胞和16细胞之间有表达变化(Hamatani et al.,2004;Wang et al., 2004)。我们注意到,对于这些数据,MPSSC在所有三个评估标准中都优于其他方法,并且所考虑的方法都没有明确区分8细胞和16细胞群体。
5 Discussion
本文介绍了一种新的谱聚类算法,该算法在目标矩阵上施加了特定的结构,其动机是观察到目标矩阵在理想情况下应该具有这种结构。我们期望施加理想的结构可以帮助获得更好的聚类结果,特别是当观测数据包含高水平的噪声和许多缺失值时。从各种模拟和单细胞数据分析中,我们看到与其他聚类方法相比,我们的算法性能有所提高。利用多个相似矩阵的扩展谱聚类算法在不同密度和不同视点的聚类中具有较好的聚类效果。在这种情况下对所提出的聚类方法进行理论分析将是我们未来的工作。在理论方面,也可以尝试第3章中的一步谱聚类方法,它的形式更简单。利用嵌入式ADMM算法迭代求解该非凸问题,并证明了算法的收敛性。只有当c > 0时,算法才会收敛,使用合适的c > 0比c=0时聚类效果更好。在不添加涉及c的项的情况下处理算法的收敛性的主题将是有趣的。虽然我们已经证明,找到 λ \lambda λ 和 μ \mu μ 的有效值通常并不难,并且在一定范围内改变这些值不会对许多聚类问题的结果产生很大影响,但我们希望这些参数的最优值应该取决于数据,而选择这些参数的数据驱动方法将是我们感兴趣的。

















 299
299

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









