








1. snakemake简介
目标:流程管理工具,创建reproducible和scalable的数据分析。
特点:
- a human readable, Python based language.
- 可以无缝地拓展到服务器、集群、网格和云环境,不需要修改工作流定义。
- 可包含所需软件的描述,这些软件可以自动部署到任何执行环境中。
1.1 初识snakemake
参考:https://snakemake.github.io/
| 特点 | 解释 |
|---|---|
| Readability(可读性) | 易于阅读、适应性强、功能强大。每个规则描述了分析中的一个步骤,该步骤定义了如何从输入文件获取输出文件。规则之间的依赖关系是自动确定的。 |
| Portability(便捷性) | 通过整合conda包管理工具和container虚拟化技术,所有软件依赖项在执行时都可以自动部署。 |
| Modularization(模块化) | 通过整合script和jupyter notebook。很容易创建和使用可重复的工具包装器,并将数据分析分解为良好分割的模块。 |
| Transparency(透明化) | 自动的,交互式的,自包含的报告确保了从结果到使用的步骤、参数、代码和软件的完整透明性。 |
| Scalability(可拓展性) | 工作流可以无缝地从单核拓展到多核、集群或云,不需要修改工作流定义和自动避免冗余计算。 |
2. Snakemake教程
链接:https://snakemake.readthedocs.io/en/stable/tutorial/tutorial.html#tutorial
Snakemake提供的符号拓展,维持了流程定义的属性。
Snakemake整合了包管理工具和容器工具Singularity,使得定义软件堆栈变成了软件的流程的一部分。
2.1安装
安装snakemake需要以下软件:
Python ≥3.5
Snakemake ≥5.24.1
BWA 0.7
SAMtools 1.9
Pysam 0.15
BCFtools 1.9
Graphviz 2.42
Jinja2 2.11
NetworkX 2.5
Matplotlib 3.3
本地安装:
wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh
bash Mambaforge-Linux-x86_64.sh ##注意是否加入PATH时,选yes。
mkdir snakemake-tutorial
cd snakemake-tutorial
##开始着手创建流程。
##首先下载示例数据。
$ wget https://github.com/snakemake/snakemake-tutorial-data/archive/v5.24.1.tar.gz
$ tar --wildcards -xf v5.24.1.tar.gz --strip 1 "*/data" "*/environment.yaml"
##创建独立的虚拟环境
$ conda activate base
$ mamba env create --name snakemake-tutorial --file environment.yaml
$ conda install -n base -c conda-forge mamba
$ conda activate snakemake-tutorial
$ snakemake --help
$ conda deactivate
报错:
ERROR Could not write out repodata file
'/share/inspurStorage/home1/jialh/mambaforge/pkgs/cache/602d3932.json': No such file or directory
解决办法:https://zhuanlan.zhihu.com/p/380567007
经多次尝试: rm -rf /Users/Likey/ProgramFiles/mambaforge/pkgs/cache/*
删除缓存文件,再次安装即可,再次安装时最好开启外网或者配置国内源环境避免其它问题.
2.2 Basic: An example workflow
Snakemake流程通过Snakefile中的特定rules来定义。Rules可以将workflow分解为small steps,通过指定如何从input files集合创建output files集合。
下面的流程来自genome analysis领域。本教程不需要你知道genome analysis。但是我们将在接下来的段落中提供一些背景。
2.2.1 Step 1: Mapping reads
Snakemake文件内容如下
rule bwa_map:
input:
"data/genome.fa",
"data/samples/A.fastq"
output:
"mapped_reads/A.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
运行方式为:
snakemake --cores 4 mapped_reads/A.bam
无论何时执行工作流程,你都可以指定使用的cores数目。本教程,我们只使用单个核。接下来,你将会看到并行化工作。注意,执行完上述命令后,Snakemake将不会再次创建mapped_reads/A.bam。 Snakemake only re-runs jobs if one of the input files is newer than one of the output files or one of the input files will be updated by another job.
Step 2: Generalizing the read mapping rule
Snakemake可以generalizing rules by using named wildcards(即通配符). 仅仅使用通配符{sample}替换上述输入文件和输出文件中的A即可。
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
Step 3: Sorting read alignments
对于后续步骤,我们需要对读段比对BAM文件排序。这可以通过samtools sort命令来实现。我们可以在bwa_map下面增加如下规则:
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam"
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
该规则会从mapped_reads获取输入文件,将排序后的版本存入sorted_reads目录。注意Snakemake会自动创建缺失的目录。对于排序,samtools需要通过字符T来指定前缀。此处,我们需要通配符sample。Snakemake也可以通过wildcards对象的value属性在shell命令中获取通配符。
Step 4: Indexing read alignments and visualizing the DAG of jobs
接下来,我们需要使用samtools来对排序后的读段比对建立索引,这样我们可以快速通过读段比对到基因组上的位置,快速获取读段(reads)。如下所示:
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input}"
注意:Snakemake使用Python format mini language来形成shell命令。有时候,你必须使用花括号{}。在那种情形下,你必须避开通过双花括号来避开它们,例如:ls {{A, B}}.txt。
共有三个步骤,可以创建结果的有向无环图(directed acyclic graph,DAG)。 可以执行:
$ snakemake --dag sorted_reads/{A,B}.bam.bai | dot -Tsvg > dag.svg
创建visualization of the DAG可以通过Graphviz的dot命令来实现。对于给定的目标文件,Snakemake可以用dot语言指定DAG,将其输入dot命令, 这可以被渲染为SVG格式。渲染的DAG会保存到dag.svg文件,如下图所示:
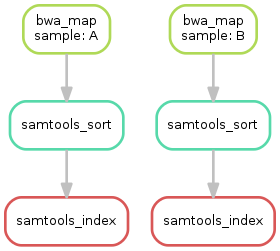
Step 5: Calling genomic variants
接下来,我们将所有mapped reads聚合到一起,联合call genomic variants。对于the variant calling, 我们将结合两个工具samtools和bcftools。Snakemake提供了helper function for collecting input files, 能够帮助我们来描述这一步的聚合。使用:
expand("sorted_reads/{sample}.bam", sample=SAMPLES)
我们将获取一个文件列表,其中"sorted_reads/{sample}.bam"模式会通过给定的sample列表SAMPLES进行格式化。例如:
["sorted_reads/A.bam", "sorted_reads/B.bam"]
这个功能对于包括多个通配符的模式非常有效,例如:
expand("sorted_reads/{sample}.{replicate}.bam", sample=SAMPLES, replicate=[0, 1])
将使用所有元素SAMPLES和列表[0, 1]来创建结果,得到:
["sorted_reads/A.0.bam", "sorted_reads/A.1.bam", "sorted_reads/B.0.bam", "sorted_reads/B.1.bam"]
此处,我们仅使用expand的简单形式。我们首先让Snakemake知道我们想考虑什么。记住,Snakemake从结果文件回溯,而不是从输入文件。因此,它不推断所有可能的输出形式,例如数据目录中的fastq文件。也要记住,Snakemake主要是增强了声明性语句的Python代码,来定义工作流。因此,我们可以在Snakemake的顶部用普通的Python特别定义的sample列表。
SAMPLES = ["A", "B"]
接下来,我们可以从config file中学习更多更复杂的形式。但是现在这足以让我们增加下面的规则到Snakefile中:
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=SAMPLES),
bai=expand("sorted_reads/{sample}.bam.bai", sample=SAMPLES)
output:
"calls/all.vcf"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
使用多个输入和输出文件,有时在shell命令中分别引用它们是很方便的。这可以通过specifying names for input or output files来实现,例如用fa=...。这些文件能够被shell通过名字引用,例如使用{input.fa}。对于long shell commands, 建议split the string over multiple indented lines。python可以自动将其合并。 进一步,你将注意到input or output file list can contain arbitary Python statements, 只要其放回字符串,或者字符串列表。此处,我们用expand函数来聚合所有样本比对上的reads。
注意:当 splitting long shell commands across multiple lines, make sure to include whitespace at the end of each line. 因为每一行的字符串只是简单地连接在一起,否则可能导致错误。例如,不能将当前的例子分割为如下命令:
"samtools mpileup"
"-g -f {input.fa} {input.bam}"
这将会连接为:
"samtools mpileup-g -f {input.fa} {input.bam}"
导致抛出错误:
[main] unrecognized command 'mpileup-g'
练习:
生成DAG图,代码为:
snakemake --dag calls/all.vcf | dot -Tsvg > dag_vcf.svg
结果如下图所示:
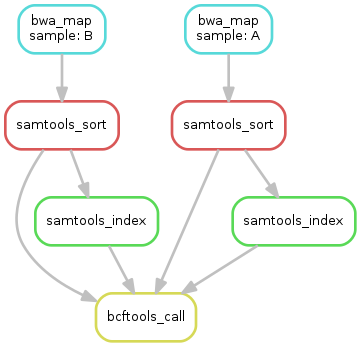
Step 6: Using custom scripts
通常,流程不仅仅是由各种工具组成,也包含手工代码来计算统计结果或者画图等等。尽管Snakemake允许你直接写python代码,但是通常将这些逻辑移到单独的脚本更合理。出于这个目的,Snakemake提供了script指令。增加如下命令到你的snakefile文件:
rule plot_quals:
input:
"calls/all.vcf"
output:
"plots/quals.svg"
script:
"scripts/plot-quals.py"
使用这个规则,我们将创建质量打分的柱状图(histogram), 其已经分配到了calls/all.vcf的变异检测中。实际上python代码将通过隐藏的脚本scripts/plot-quals.py来产生柱状图。脚本的路径通常是相对Snakefile的。在脚本中,规则的所有属性像input, output, wildcards等snakemake全局对象属性也可以使用。 使用如下内容,产生scripts/plot-quals.py文件:
import matplotlib
matplotlib.use("Agg")
import matplotlib.pyplot as plt
from pysam import VariantFile
quals = [record.qual for record in VariantFile(snakemake.input[0])]
plt.hist(quals)
plt.savefig(snakemake.output[0])
除了python代码,可以使用R代码。详情参考:https://snakemake.readthedocs.io/en/stable/snakefiles/rules.html#snakefiles-external-scripts
Step 7: Adding a target rule
到目前为止,我们总是通过指定命令行中目标文件的名字来执行流程。除了文件名,Snakemake also accepts rule names as targets,如果访问的规则不包括通配符。因此,可以编写收集所需结果的特定子集或所有结果的目标规则。而且,如果命令行没有给目标,Snakemake也可以定义Snakefile的first rule作为目标。 因此最好是在workflow顶部设置all规则,将所有目标输出文件作为输入文件。
此处,我们可以增加规则:
rule all:
input:
"plots/quals.svg"
到workflow的顶部。 然后执行Snakemake:
snakemake -n --cores 4
练习:
- 绘制完整流程的DAG图。
snakemake --dag plots/quals.svg | dot -Tsvg > dag_quals.svg
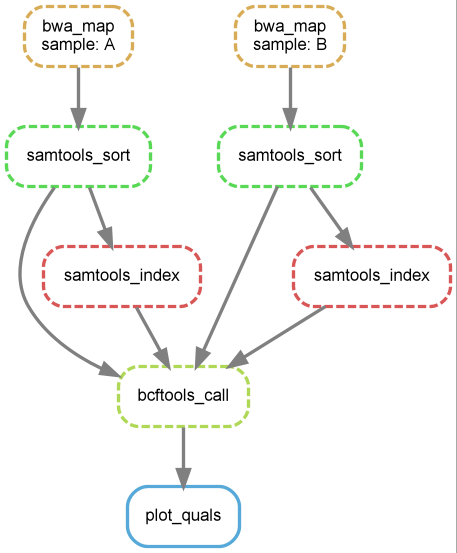
- 执行完整的流程,看一看结果
plots/quals.svg。
snakemake -n --cores 4
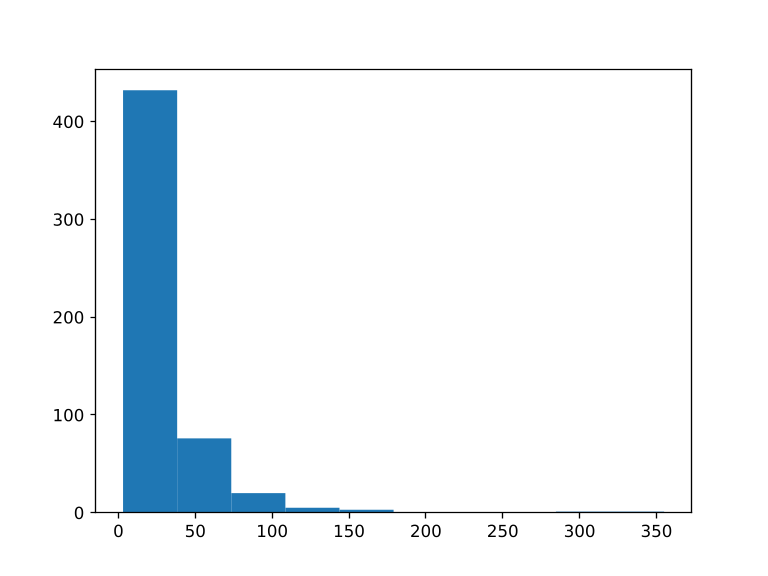
- 查看
snakemake --help, 选取部分参数,如下所示:
-h, --help ##展示帮助文档。
--dry-run, --dryrun, -n ##不执行任何东西,仅仅展示流程会干什么。
--profile PROFILE ##用于参数化Snakemake的profile名字。
--cache [RULE [RULE ...]] ##通过环境变量$SNAKEMAKE_OUTPUT_CACHE, 存储给定规则输出文件的中间cache。
--snakefile FILE, -s FILE ##snakefile形式的工作流程定义。
--cores [N], -c [N] ##最多使用多少个CPU cores/jobs用于并行化。
--jobs [N], -j [N] ##最多使用多少个CPU cluster/cloud jobs用于并行化。
--local-cores N ##在使用cluster/cloud模式时,主机使用多少个cores来并行化。
--resources [NAME=INT [NAME=INT ...]], --res [NAME=INT [NAME=INT ...]] ##定义额外的资源。
--set-threads RULE=THREADS [RULE=THREADS ...] ##重写threads规则。
--max-threads MAX_THREADS ##定义任何工作的最大threads数目。
--set-resources RULE:RESOURCE=VALUE [RULE:RESOURCE=VALUE ...] ##重写rules使用的资源。
--config [KEY=VALUE [KEY=VALUE ...]], -C [KEY=VALUE [KEY=VALUE ...]] ##设置和重写流程config对象的值。
--configfile FILE [FILE ...], --configfiles FILE [FILE ...] ##指定或重新流程config file(见文本)。
--envvars VARNAME [VARNAME ...] ##传递到cloud jobs的环境变量数。
--directory DIR, -d DIR ##指定工作路径。
--touch, -t ##touch输出文件(更新日期,不需要真正改变它们)。
--keep-going, -k ##如果一个jobs失败,与之相关的jobs是否继续执行。
--force, -f ##忽略所有输出,从第一个rule或者选定的target来强制执行。
--forceall, -F ##忽略所有输出,强制执行选定的(或第一个)rule和所有其依赖的rules。
--forcerun [TARGET [TARGET ...]], -R [TARGET [TARGET ...]] ##强制重新执行给定的规则或者产生给定的文件。使用这个选项,如果你改变了一个rule, 想要你的流程中所有它的输出都更新。
--prioritize TARGET [TARGET ...], -P TARGET [TARGET ...] ##告诉调度器为给定目标(及其所有依赖项)的创建分配最高优先级。
--batch RULE=BATCH/BATCHES ##为给定RULE的输入文件创建BATCH。
--until TARGET [TARGET ...], -U TARGET [TARGET ...] ##重新运行此rule, 直到其到达指定的rules或者files。
--omit-from TARGET [TARGET ...], -O TARGET [TARGET ...] ##防止执行的规则或者创建的文件,是DAG中这些目标的下游。
--rerun-incomplete, --ri ##重新运行所有输出被认为是不完整的任务。
使用--forcerun重新执行samtools_sort规则,看看会发生什么。
snakemake --cores 4 --forcerun samtools_sort
流程重新执行了samtools_sort及其后面的所有步骤。
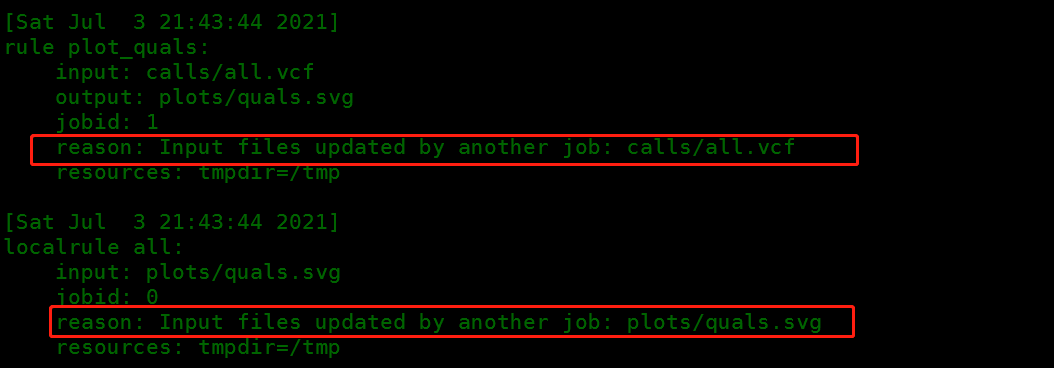
- 使用
--reason,它能够展示每个job执行的理由。尝试使用这个指令以及dry-run和--forcerun,理解snakemake的决策。
snakemake --cores 1 --forcerun samtools_sort -n --reason
执行结果,会产生reason部分,说明为什么会执行这一步。
Summary
总而言之,结果流程如下所示:
SAMPLES = ["A", "B"]
rule all:
input:
"plots/quals.svg"
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam"
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input}"
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=SAMPLES),
bai=expand("sorted_reads/{sample}.bam.bai", sample=SAMPLES)
output:
"calls/all.vcf"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
rule plot_quals:
input:
"calls/all.vcf"
output:
"plots/quals.svg"
script:
"scripts/plot-quals.py"

















 452
452

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









