







文章目录
参考:https://snakemake.readthedocs.io/en/stable/tutorial/advanced.html
进阶部分:对案例进行进一步修饰
对基础部分的案例流程进行进一步的修饰。
目前Snakemake的基本概念已经得到了展示,接下来我们将介绍一些进阶用法。
Step 1: Specifying the number of used threads
Snakemake可以使用threads指令来满足程序的threads(线程)需求。可以在bwa_map规则中使用threads命令,如下所示:
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
threads: 8
shell:
"bwa mem -t {threads} {input} | samtools view -Sb - > {output}"
threads数目的参数可以通过花括符(例如{threads})的形式传递到shell命令。如果没有给定threads指令,那么默认该rule使用1个thread。
Snakemake会统筹使用的threads总数,不会超过命令行中--cores指令的规定,其在snakemake执行命令时传递。例如:
$ snakemake --cores 10
将会用10个cores来执行流程。由于bwa_map需要8个threads, 因此一次只能运行一个job, 剩下的cores会被用来执行其他工作,例如samtools_sort。rule中的threads指令可以被解释为最大threads, 如果能提供的threads较少,threads将会减少到给定的cores数目。
注意:处理threads, snakemake还提供了resource来控制内存、GPU等资源(详情见resource)。
练习
使用--forceall强制重新运行流程,集合--cores的不同值,检查流程会如何选择工作来并行化运行。
snakemake --cores 16 --forceall ##2个job同时执行bwa_map。
snakemake --cores 10 --forceall ##1个job执行bwa, 完成后开始执行samtools_sort。
Step 2: Config files
目前,我们通过Snakefile中的Python列表来指定考虑哪些样本。然而,通常你需要你的流程是可制定的,进而能够适应新的数据。出于此目的,Snakemake提供了config file mechanism。Config文件可以被写为JSON或YAML形式。在我们的示例流程中,我们可以增加如下行到Sankefile的顶部:
configfile: "config.yaml"
Snakemake会加载contig文件,然后将其内容存储为一个全局字典,名为config。此处,我们可以在config.yaml中指定samples。
samples:
A: data/samples/A.fastq
B: data/samples/B.fastq
现在,我们可以删掉Snakefile中的SAMPLES, 并改变bcftools_call规则为:
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=config["samples"]),
bai=expand("sorted_reads/{sample}.bam.bai", sample=config["samples"])
output:
"calls/all.vcf"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
运行:
snakemake --cores 16 --forceall
出现了警告信息:[warning] samtools mpileup optiongis functional, but deprecated. Please switch to using bcftools mpileup in future. [mpileup] 2 samples in 2 input files.
解决办法:
将samtools mpileup -g -f改为bcftools mpileup -f。
Step 3: Input functions
由于我们已经在confige文件中存储了FASTQ文件的路径,我们也可以用这些路径泛化bwa_map规则。这种情形不同于我们上面修改过的bcftools_call。为了理解这个,知道Snakemake流程在三个阶段的执行很重要。
- 初始化阶段:定义workflow的文件被解析,所有rules被实例化。
- DAG阶段:通过填充所有通配符,匹配输入和输出文件,构建所有工作的有向无环依赖图。
- scheduling阶段:工作的DAG执行后,根据所有可用的资源开始工作。
初始阶段,不会执行bcftools_call中规则输入文件列表中的拓展函数,因而我们不能够确定bwa_map规则中的FASTQ的路径。相反,我们在DAG阶段确定输入文件。这可以通过input函数,而不是input directive中的字符串。对于bwa_map规则:
def get_bwa_map_input_fastqs(wildcards):
return config["samples"][wildcards.sample]
rule bwa_map:
input:
"data/genome.fa",
get_bwa_map_input_fastqs
output:
"mapped_reads/{sample}.bam"
threads: 8
shell:
"bwa mem -t {threads} {input} | samtools view -Sb - > {output}"
任何正常的函数工作得都很好。输入函数接收一个wildcards对象作为single argument,这允许通过属性(此处是wildcards.sample)来获取wildcards的值。 他们必须要返回一个字符串或者字符串列表,其可以被解释为输入文件的(此处我们返回config文件中包含的sample路径)。一旦一个工作的wildcards值被确定,输入函数就可以被评估。
练习:
将data/samples目录的C.fastq增加到config文件中,然后执行如下命令,看看snakemake如何重新计算流程中的属于新样本的部分。
snakemake -n --reason --forcerun bcftools_call --cores 4
Step 4: Rule parameters
有时候,shell命令不一定由input和output文件以及某些静态标记组成。特别地,需要根据jobs中的通配符来设置额外的参数。对此,Snakemake允许rule通过params指令difine arbitary parameters(定义经验参数)。在我们的流程中,合理的做法是将比对上的读段和所谓的读段组进行注释,那么可以在metadata中包含sample name。我们可以将bwa_map规则修改为:
rule bwa_map:
input:
"data/genome.fa",
lambda wildcards: config["samples"][wildcards.sample]
output:
"mapped_reads/{sample}.bam"
params:
rg=r"@RG\tID:{sample}\tSM:{sample}"
threads: 8
shell:
"bwa mem -R '{params.rg}' -t {threads} {input} | samtools view -Sb - > {output}"
类似于输入和输出文件,params也可以通过shell命令、基于run模块的Python或者script指令来获取。
练习
Variant calling会考虑很多参数。非常重要的是先验突变率(默认为$1e-3$)。可以通过bcftools call命令的-P参数来设置。 考虑如何在config文件中增加新的key,然后使用bcftools_call规则中的params指令,将其传递给shell命令。
修改后运行如下命令,顺利运行。
snakemake --forcerun bcftools_call --cores 4
Step 5: Logging
当执行一个大的流程时,理想的是每个job的logging output输出到一个单独的文件,而不是将所有logging output都输出到terminal–当多个jobs并行化运行时,会导致混乱的输出。出于这个目的,Snakemake允许为rules指定log file。 Log file通过log指令来定义,类似于output文件,但是它们不受规则约束,当作业失败时,也不会清除它们。我们可以将bwa_map规则修改为如下:
rule bwa_map:
input:
"data/genome.fa",
get_bwa_map_input_fastqs
output:
"mapped_reads/{sample}.bam"
params:
rg=r"@RG\tID:{sample}\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
threads: 8
shell:
"(bwa mem -R '{params.rg}' -t {threads} {input} | "
"samtools view -Sb - > {output}) 2> {log}"
Shell命令可以被bwa和samtools的collect STDERR output修改,然后将其输入{log}指定的文件。 Log files必须包括与输出文件相同的通配符来避免相同规则的不同jobs的file name冲突。
练习
(1)尝试给bcftools_call增加log指令。
(2)重新运行整个程序流程,查看variant calling和read mapping步骤如何创建log文件。
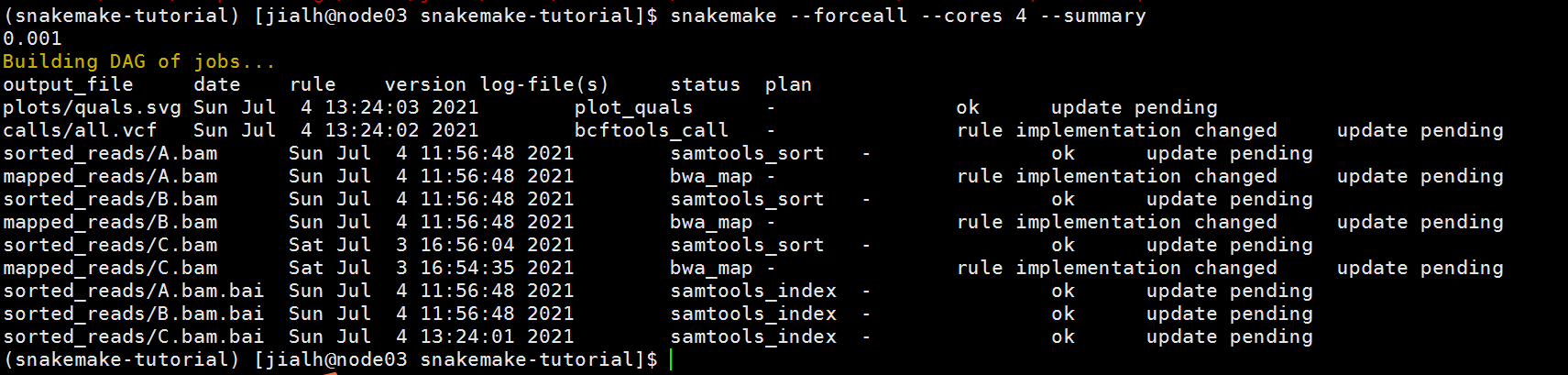
(3)记录产生结果相关的重要步骤,有助于重复分析。上面讨论的report, Snakmake能够流程输出文件的各种相关信息。而--summary能够打印一个表格,将每个输出文件与用于生成它的规则、创建日期以及用于创建的工具(可选)版本关联起来。而且,改表格还能够提供输出文件创建后的输入文件更新、源代码改变等信息。使用带--summary的Snankemake,能够检查我们的案例中的信息。
尝试执执行一下:
snakemake --forceall --cores 4 --summary
结果如下所示:
Step 6: Temporary and protected files
在上述流程中,我们为每个样本创建了两个BAM文件,分别是bwa_map以及samtools_sort的输出。当不是处理示例数据时,实际的数据是非常大的。因此结果BAM文件也非常大,需要很多存储文件,创建也需要花费很多时间。为了节省磁盘空间,你可以将某些输出指定为临时文件(mark output files as temporary)。一旦需要其作为输入文件的流程全部执行完了, Snakemake能够为你删掉标记的文件。我们为bwa_map规则的输出执行使用这个机制:
rule bwa_map:
input:
"data/genome.fa",
get_bwa_map_input_fastqs
output:
temp("mapped_reads/{sample}.bam")
params:
rg=r"@RG\tID:{sample}\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
threads: 8
shell:
"(bwa mem -R '{params.rg}' -t {threads} {input} | "
"samtools view -Sb - > {output}) 2> {log}"
一旦对应的samtools_sort工作执行结束,BAM文件就会被删掉。因为BAM是通过读段比对和排序得到的,计算消耗非常大,因此保护最终的BAM文件放置误删或者修改(from accidental deletion or modification)。我们可以将samtools_sort的输出标记为protected类型。
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
protected("sorted_reads/{sample}.bam")
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
尝试执行代码:
snakemake --forceall --cores 4
结果发现,mapped_reads目录不存在,而sorted_reads目录下的{A,B,C}.bam变为了只读权限。
练习
-
重新执行整个流程,观察Snakemake是如何处理temporary和protected文件。
-
运行Snakemake使用目标
sorted_mapped/A.bam。可以发现,尽管该文件被标记为temporary文件,但是Snakemake不会删掉它,因为它被指定为target file。snakemake --cores 4 mapped_reads/A.bam -
尝试使用dry-run选项,重新执行整个流程。你将会发现,其会失败,因为Snakemake不会重新写保护的输出文件。
snakemake -n --cores 4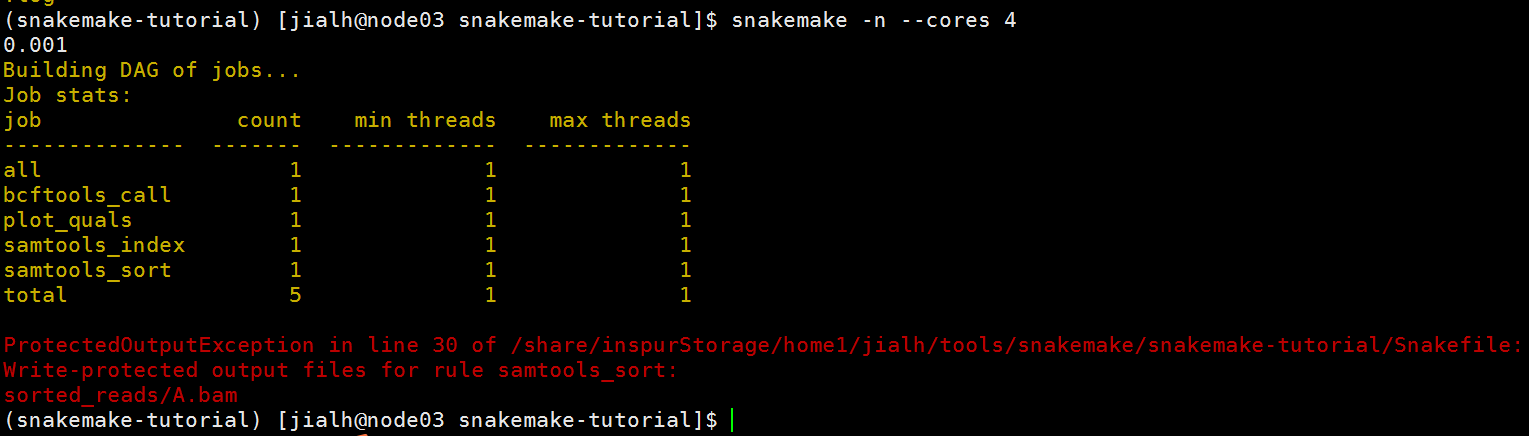
Summary
作为本教程的进阶部分,你现在学会了如何创建contig.yaml配置文件:
samples:
A: data/samples/A.fastq
B: data/samples/B.fastq
prior_mutation_rate: 0.001
使用这个文件,最终的Snakefile工作流程如下所示:
configfile: "config.yaml"
rule all:
input:
"plots/quals.svg"
def get_bwa_map_input_fastqs(wildcards):
return config["samples"][wildcards.sample]
rule bwa_map:
input:
"data/genome.fa",
get_bwa_map_input_fastqs
output:
temp("mapped_reads/{sample}.bam")
params:
rg=r"@RG\tID:{sample}\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
threads: 8
shell:
"(bwa mem -R '{params.rg}' -t {threads} {input} | "
"samtools view -Sb - > {output}) 2> {log}"
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
protected("sorted_reads/{sample}.bam")
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input}"
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=config["samples"]),
bai=expand("sorted_reads/{sample}.bam.bai", sample=config["samples"])
output:
"calls/all.vcf"
params:
rate=config["prior_mutation_rate"]
log:
"logs/bcftools_call/all.log"
shell:
"(samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv -P {params.rate} - > {output}) 2> {log}"
rule plot_quals:
input:
"calls/all.vcf"
output:
"plots/quals.svg"
script:
"scripts/plot-quals.py"















 228
228











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









