





VASP 6.3 机器学习力场
前言
VASP 6.3 正式发布了机器学习力场功能,在IBRION=0 进行从头算分子动力学模拟(AIMD)时只需在INCAR中控制两个标签ML_LMLFF ;ML_ISTART便能在跑分子动力学时选择是否引入即时(on-the-fly)机器学习力场。官方宣称在从头算分子动力学模拟中引入on-the-fly机器学习力场将会显著降低模拟时间。本文将对VASP官方文档中新增机器学习力场进行介绍。想直接看实操视频的同学可以关注B站up主‘冬夜秋雾’的视频。或者官方文档的算例
一、 VASP 机器学习力场:理论
介绍
分子动力学是研究材料动力学性质的重要方法。分子动力学模型的精度严重依赖计算方法,高精度的结果常常以高计算成本为代价。从头算分子动力学方法具有高精度的优势,原因在于原子间的相互作用是通过从头算方法计算的,例如基于密度泛函的从头算(DFT)。不幸的是,该方法常被限制在很小的晶胞和模拟时间之内,且需要很长的计算时间。可以加速计算时间的一种方法是使用力场,它是势能的参数化形式。 这些力场可以是基于经验开发的简单函数,也可以是基于上千次DFT计算结果拟合的非常复杂的函数。通常手工制作精确的力场是一项非常耗时的任务,需要大量的专业知识和技术。
另一种可以降低计算耗时与人工参与的方法是机器学习。它可以基于已经完成的DFT计算结果上进行自动插值。与需要手动(甚至经验)调整参数的经典力场相比,这种方法已经显著简化了力场的生成。然而,如何选择合适的数据进行训练仍旧是个问题。一种非常有效的解决办法是进行‘即时学习’(on-the-fly,或者翻译成动态学习更好)。其原理是,在进行从头算分子动力学模拟时会随时产生已经算好的数据,经过一些算法判断,选择需要的数据进行机器学习操作,也就是说,建立原子位置与能量、力、或许还有应力的映射关系。这样就建立起了机器学习力场。其中的关键点,也是它被称为动态学习的核心,在于判断每一步计算(解牛顿第二定律方程)是使用已建立的机器学习力场作为势函数,还是使用DFT计算结果。
这个判据是通过机器学习算法给出的,在VASP中,使用的是贝叶斯算法来拟合原子位置与力的关系,所以可以用贝叶斯误差来估计真实的预测误差。(这里需要特别注意,官网也着重强调过,贝叶斯误差是对预测误差的估计,为什么是估计值,因为当我们开始一个新步计算时,当前晶胞的能量、力这些信息我们是不知道的,因为我们还没算出来,此时可以通过机器学习力场预测出能量和力,但由于不知道真实值是多少,也就没有办法计算预测的误差,贝叶斯算法相比于其它机器学习算法的一个特点就是,它可以对预测误差进行估算,当然其中的原理可以搜索贝叶斯算法进行了解,我们只需要知道在VASP的on-the-fly学习中,测试误差都是用误差的估计来近似的)。在有了预测误差的估计后,on-the-fly便可以通过设定的误差阈值来判断当前步是直接用机器学习力场预测还是DFT计算。到这儿,基本就了解了为什么说加入机器学习力场后,分子动力学的计算速度会大大提升,因为并不是每一步都进行了DFT计算,我们可以简单的把这种方法理解为:有些步用的从头算分子动力学,有些步用的类似于传统势函数的分子动力学,所以当然比纯用从头算分子动力学的方法快。并且,由于是利用机器学习模型来进行的力场拟合(势函数拟合),这比传统势函数预测的更加精准,在一定条件下完全可以和DFT计算的精度相当,所以,这种on-the-fly方法即节省计算时间,又不会牺牲太多预测精度,可以说是一举两得。
流程
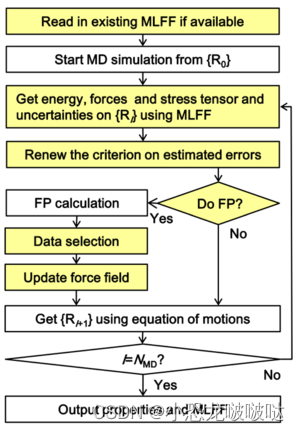
这里我结合自己的理解解释一下流程:
1.开始第一步从头算分子动力学计算
2.在进行若干步从头算分子动力学后,利用已经算出来的晶胞构型和能量、力、应力的数据建立机器学习模型,也就是机器学习力场。
3.开始进入上图中的循环:
4. 利用机器学习力场对当前晶胞的能量,力,应力进行预测
5. 判断预测误差的估计是否在阈值内,如果在则利用机器学习力场进行当前分子动力学模拟(解牛顿方程)并从第3步循环,如果不在则进行DFT计算(解K-S方程),得到当前晶胞的能量、力、应力。
6. 将当前DFT数据放入待训练区,等待积攒到一定数据量后加入到已有的训练集从头训练新的机器学习力场(主动学习)
7. 判断模拟步数是否达到预设值,到达即结束分子动力学模拟,输出最终机器学习力场模型,未达到从第3步开始循环.
上面的流程是简化了的,实际的on-the-fly算法还有许多细节,比如对当前步是否该进行DFT计算的判断,我简化成了直接判断预测误差的估计是否在阈值内,实际上官方的算法更复杂,判断条件更多,其目的就是尽可能的保证力场精度和计算时间之间的平衡,更详细的可以到官网查看。
缺点
机器学习领域有一句经典名言“天下没有免费的午餐”。分子动力学是对物理世界的模型,真实物理世界的每一个现象都是多因素的影响结果,当我们提升了模拟时间后,不可避免地会牺牲一部分东西。在机器学习力场上,牺牲的东西就是力场的迁移性。
想象一下,使用VASP的on-the-fly算法,理论上,只要一直模拟下去,最终我们将得到一个可以包含所有元素,所有晶体构型的机器学习力场,到时候我们将可以在分子层面模型物理世界的任何情况。但这确又并不现实。
所谓迁移性,就是模型在训练数据以外的晶体构型上的预测能力。官网举了一个很典型的例子,当新构型晶胞的密度在训练数据中晶胞的密度范围内时,预测结果会很理想(预测构型和训练数据构型元素类型相同)。这很容易理解,两者密度相似,说明构型在某种程度上相似,机器学习模型的本质是插值,这种情况当然就会插值准确。但是一旦预测构型与训练数据中的相差太远,其预测结果将会偏离严重。可以用一篇综述中的图来生动表示
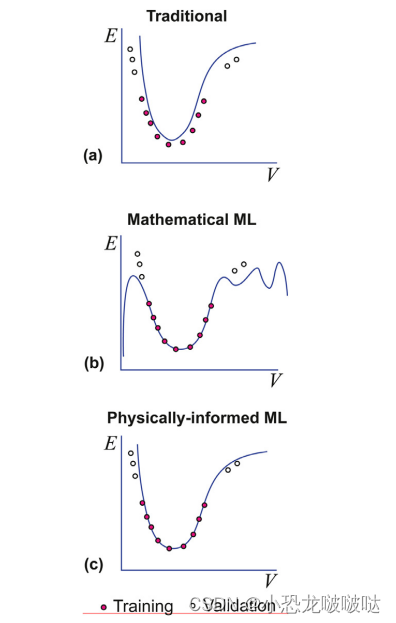
a b c 依次为传统势函数、机器学习势函数、物理信息加持的机器学习势函数的外推预测能力。可以看到,经典势函数虽然拟合精度较差,但由于它是基于物理规律拟合的公式,所以其在其在训练点之外的外推预测能力还算可以,至少不会像机器学习力场那样离谱(虽然训练点拟合得非常完美)。
这里有一种新的机器学习力场,基于物理信息的机器学习力场,该力场是对机器学习方法和传统势函数的结合,简单说就是通过机器学习来拟合传统势函数中的参数,再用拟合的参数构建传统势函数,这样既可以简化势函数的开发,又可以保证势函数的外推能力。目前这种方法还在进一步探索中。
回到VASP的机器学习力场中,该力场是纯的机器学习力场,也就是上图b中的情况,其外推能力是很有限的,简言之,如果我用CuO的机器学习力场去预测ZnO的能量、力信息,那一定是不准确的,我用bulk训练的力场去预测界面的受力信息,那大概率也是误差巨大。
以上是对VASP机器学习力场的介绍和部分见解,因为才接触这一块,所以可能会有一些错误,欢迎大家纠错。

















 1102
1102

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









