







常见的单细胞转录组图形基本都包含,用起来也比较方便,很适合不擅长生信分析的人来做数据探索。对于做分析的人来讲,这样一个网页工具也降低了和非分析合作者的沟通成本。
该工具2019年发表在Bioinformatics,文献标题"Cerebro: interactive visualization of scRNA-seq data".
具体来讲,Cerebro的功能主要包括:
- 交互式降维结果展示
- 差异基因展示
- 通路富集分析结果展示
- 基因和基因集评分的展示
- 拟时序分析结果展示,用的Monocle2
- 导出图片为pdf
- 表格的导出
Cerebro的使用方法也很简单,首先需要准备好一个较完整的seurat对象,细胞注释这一步最好已经完成。得到这样一个seurat对象的代码为1.seurat.R,本文不演示。(公粽号上)本文最后会提供链接,用于获取本文涉及到的所有代码和示例数据。
下面先来看一下,如何生成这个shiny工具需要的.crb文件。
1. 安装R包并加载
cerebroApp这个R包是配套Cerebro工具使用的,这个R包的目的主要是准备Cerebro的输入文件,也包括一些基本分析,比如找差异基因、富集分析,都是一步搞定的。详细教程在https://romanhaa.github.io/cerebroApp/articles/cerebroApp_workflow_Seurat.html
BiocManager::install('romanhaa/cerebroApp')
library(cerebroApp)
2. 导入seurat对象,并进行基本分析
testEC=readRDS("testEC.rds")
2.1 计算线粒体、核糖体基因表达占比
testEC <- addPercentMtRibo(testEC,organism = 'hg',gene_nomenclature = 'name')
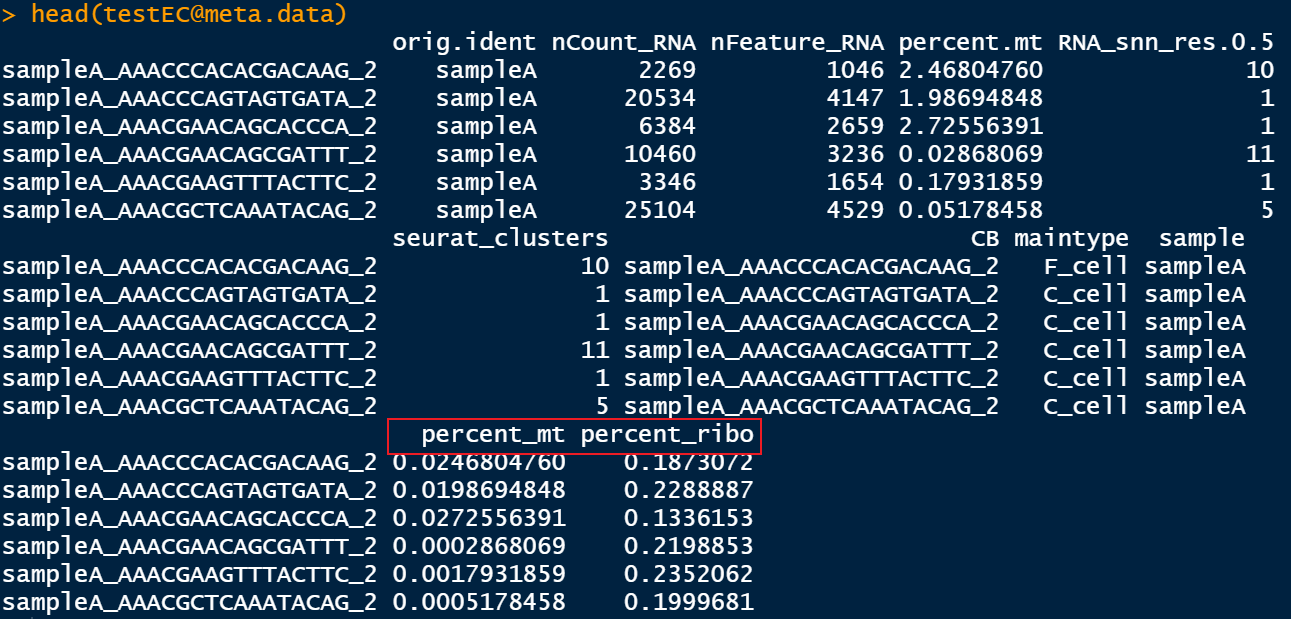
之后meta数据框会多出两列。此外,很神奇的是,所用到的线粒体、核糖体基因集会被存储在:
testEC@misc$gene_lists$mitochondrial_genes
testEC@misc$gene_lists$ribosomal_genes
我也是头一回知道seurat对象还可以有这个misc slot, 用于存储一些有用的基因集、数据框。
2.2 获取表达最多的基因
这里表达最多是从UMI总数的角度来说的,是指在特定的分组情况下,某个基因的UMI之和,比上所有基因的UMI之和。
testEC <- getMostExpressedGenes(testEC,assay = 'RNA',groups = c('sample','maintype'))
比如用maintype分组,就是求每种细胞类型里面,哪些基因表达最多。这些基因以数据框的形式存储:
testEC@misc$most_expressed_genes$sample
testEC@misc$most_expressed_genes$maintype
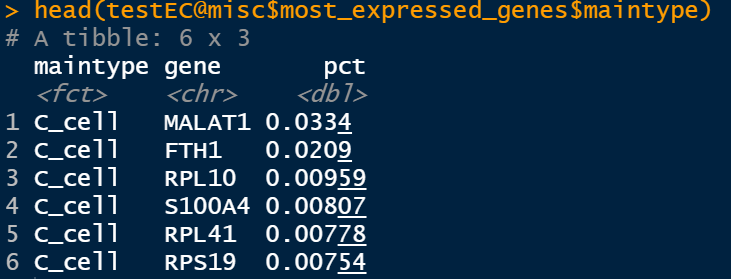
2.3 获取marker基因
这一步调用的是seurat的FindAllMarkers
testEC <- getMarkerGenes(
testEC,
assay = 'RNA',
organism = 'hg',
groups = c('sample','maintype'),
name = 'cerebro_seurat',
only_pos = TRUE,
min_pct = 0.7,
thresh_logFC = 0.25,
thresh_p_val = 0.01,
test = "wilcox"
)
结果存储在:
testEC@misc$marker_genes$cerebro_seurat$sample
testEC@misc$marker_genes$cerebro_seurat$maintype
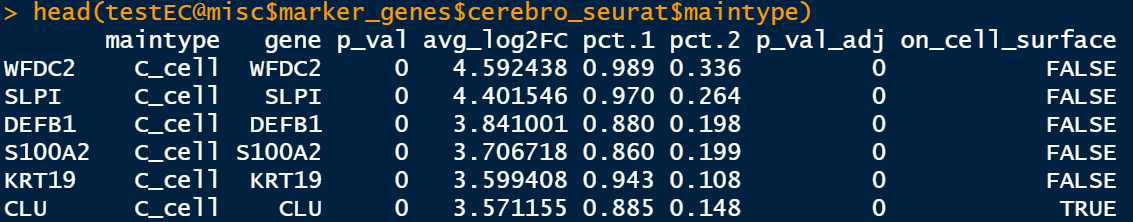
这里最后一列是on_cell_surface信息,这个seurat里面好像是没有的。
2.4 通路富集分析
testEC <- getEnrichedPathways(
testEC,
marker_genes_input = 'cerebro_seurat',
adj_p_cutoff = 0.01,
max_terms = 100
)
结果以数据框的形式存储在:
testEC@misc$enriched_pathways$cerebro_seurat_enrichr$sample
testEC@misc$enriched_pathways$cerebro_seurat_enrichr$maintype
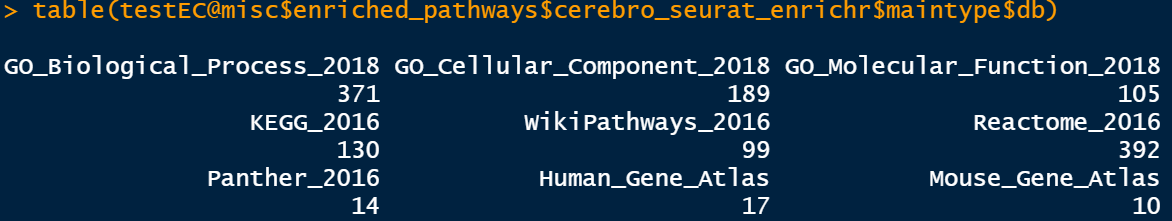
这一步富集用到的数据库比较全,包括:
2.5 GSEA也能做
不过只是得到表格,不出图
gmt_path <- "D:/hsy/bioinformatics/PTJ/019_EC_600/metabolic_pathways3.gmt"
testEC <- performGeneSetEnrichmentAnalysis(
testEC,
assay = 'RNA',
GMT_file = gmt_path,
groups = c('sample','maintype')
)
# 结果存储在:
# testEC@misc$enriched_pathways$cerebro_GSVA$sample
# testEC@misc$enriched_pathways$cerebro_GSVA$maintype
3. 导出crb文件
exportFromSeurat(
testEC,
assay = 'RNA',
slot = 'data',
file = 'testEC.crb',
experiment_name = 'EC',
organism = 'hg',
groups = c('sample','seurat_clusters','maintype'),
nUMI = 'nCount_RNA',
nGene = 'nFeature_RNA',
add_all_meta_data = TRUE,
verbose = FALSE
)
这一步会在工作目录下面生成.crb文件
4. 打开shiny工具
launchCerebro(version = "v1.3")
之后你的浏览器就会自动打开一个网页,如下:

点击上方的Browse按键,导入上一步得到的文件,之后就可以开始可视化了
5. 图形展示
最左边的是菜单栏
5.1 降维图,对应菜单栏为overview
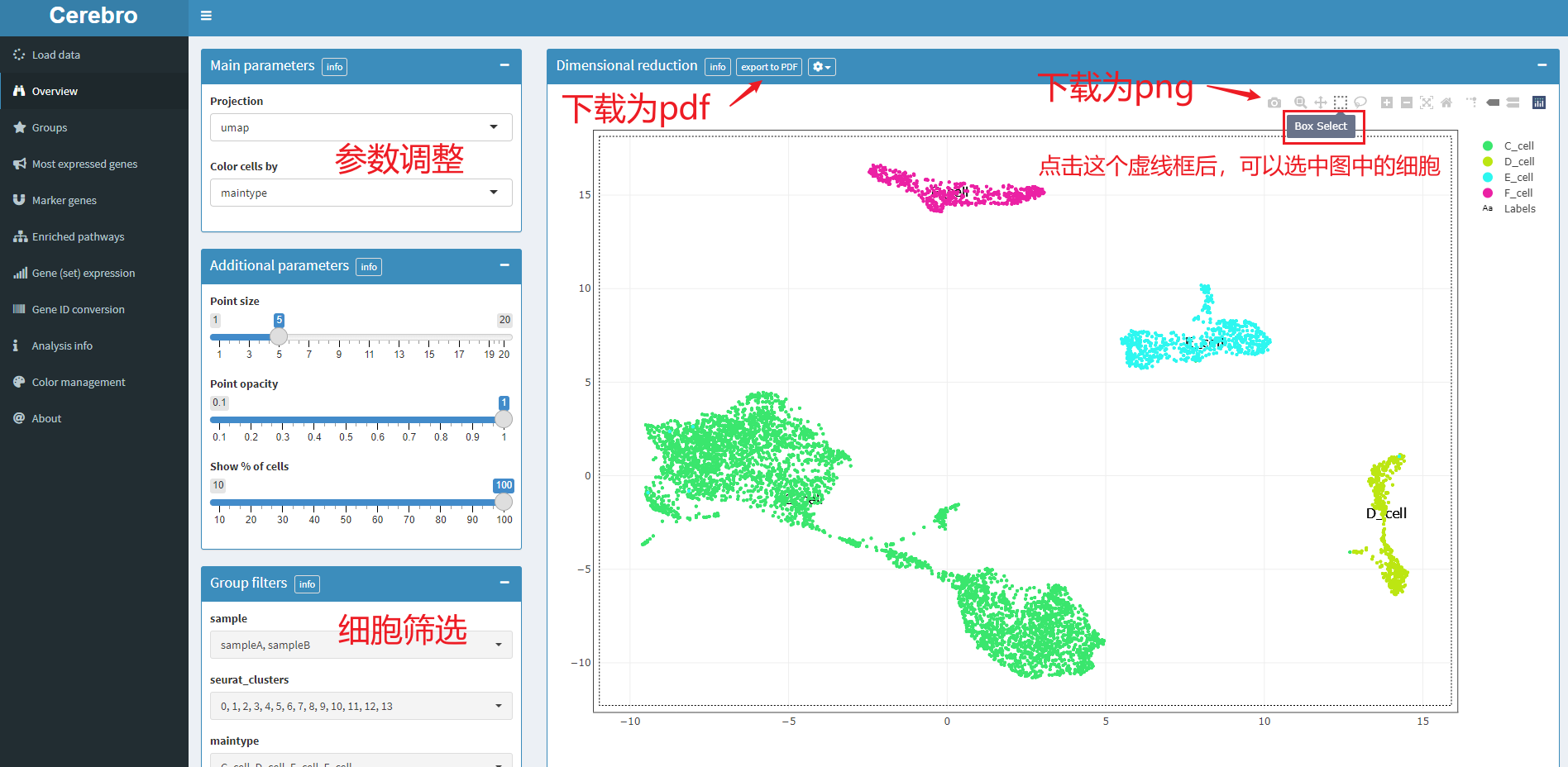
左边有一列参数调整的控件,降维方式、分组方式、点的大小和透明度、是否筛选细胞等等这些都可以调整。
右侧绘图区,可以下载图片,也可以选中细胞。选中的细胞在该页面下方会有统计信息,包括细胞数量,以及meta数据框中的属性信息。如下:
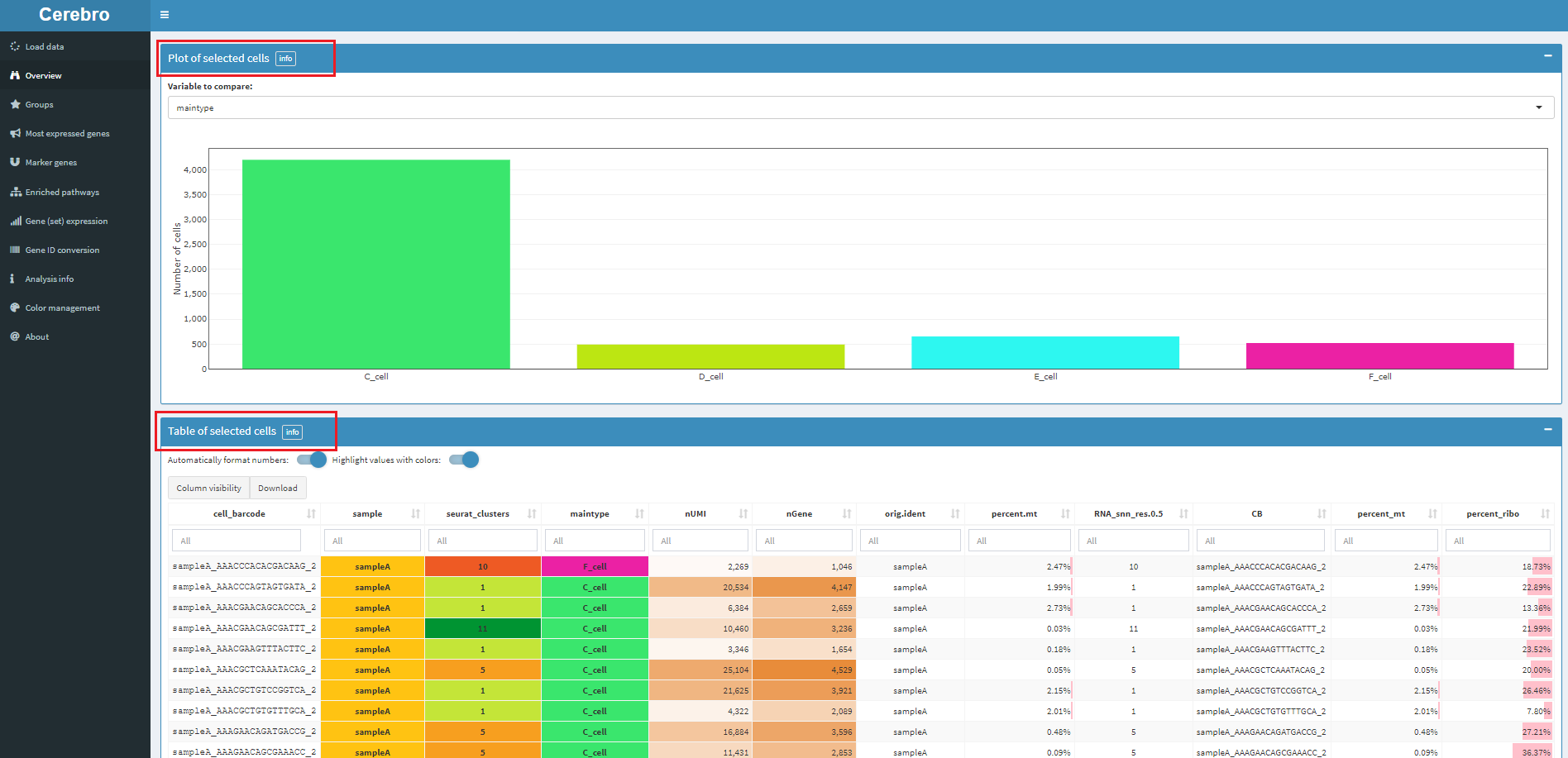
5.2 细胞类型的占比,几个质控指标
对应菜单Groups

上图为每个样本里面,各种细胞类型的占比
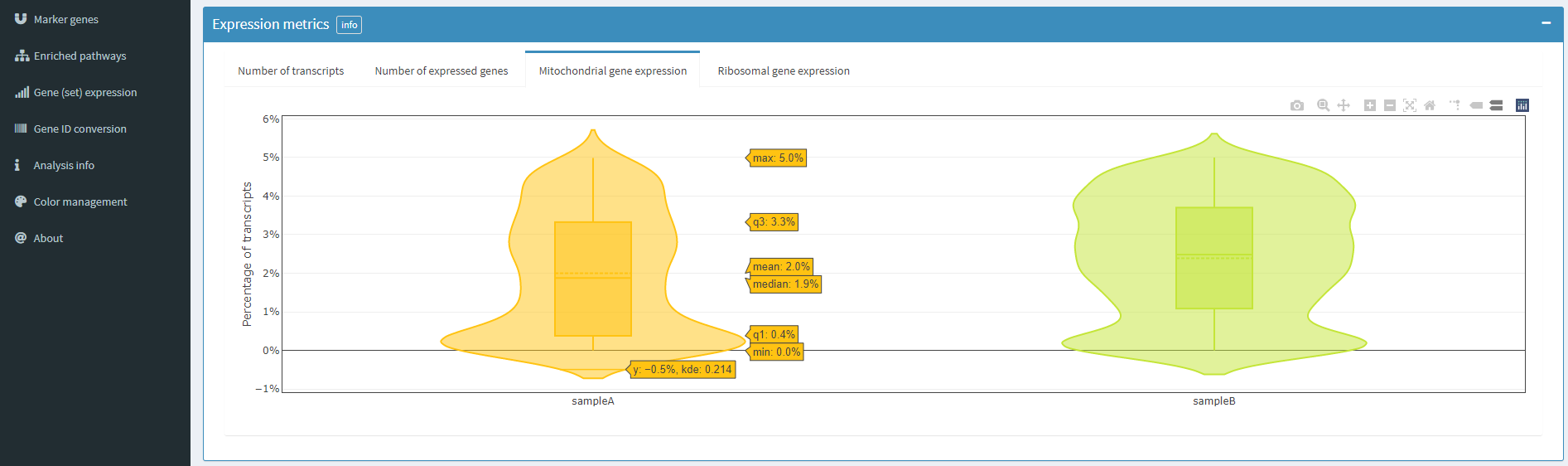
上图为每个样本线粒体基因表达占比
5.3 表达最多的基因,对应菜单栏为most expressed gene
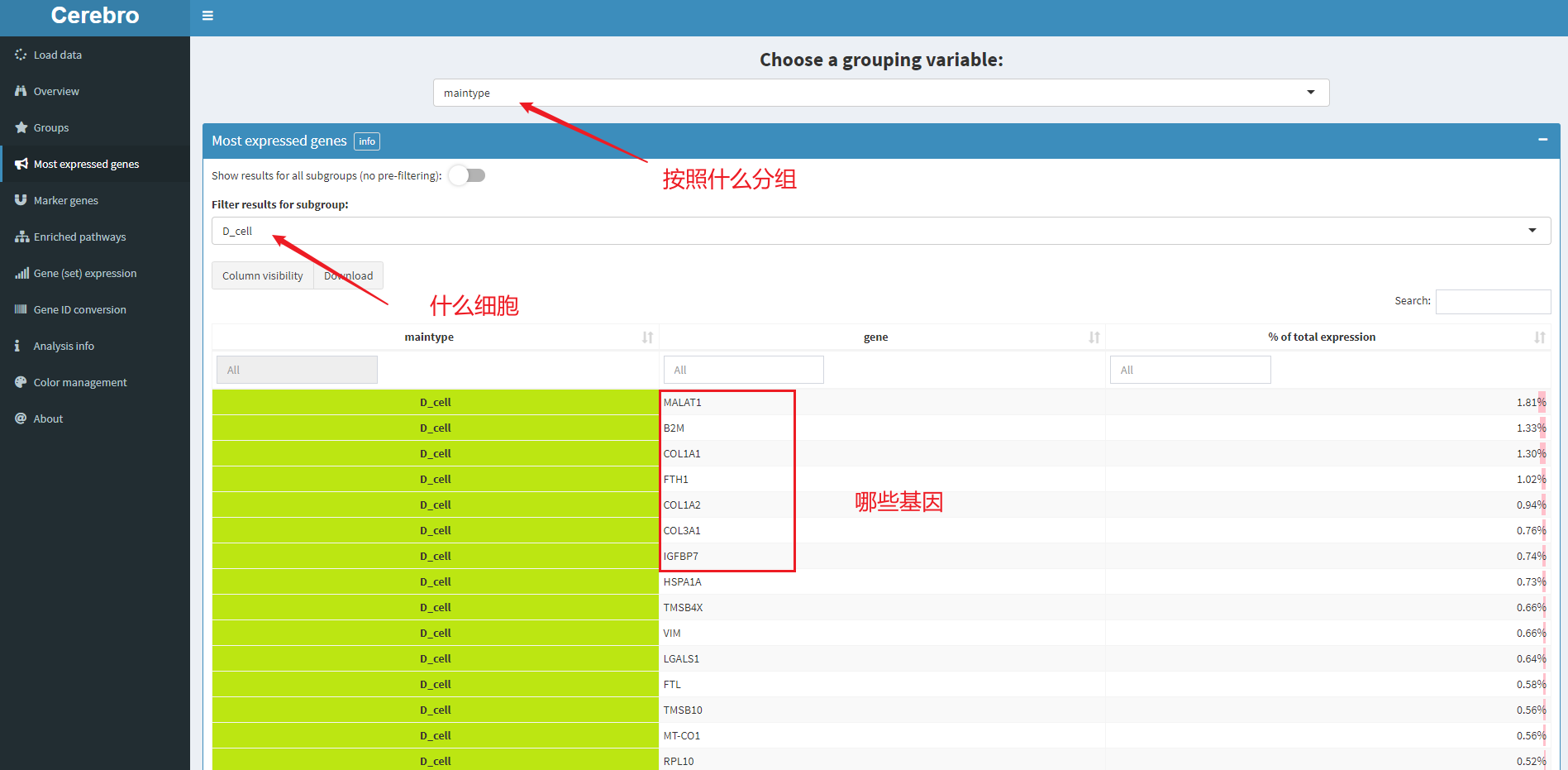
如图,表示D_cell中表达最多的基因
5.4 差异基因
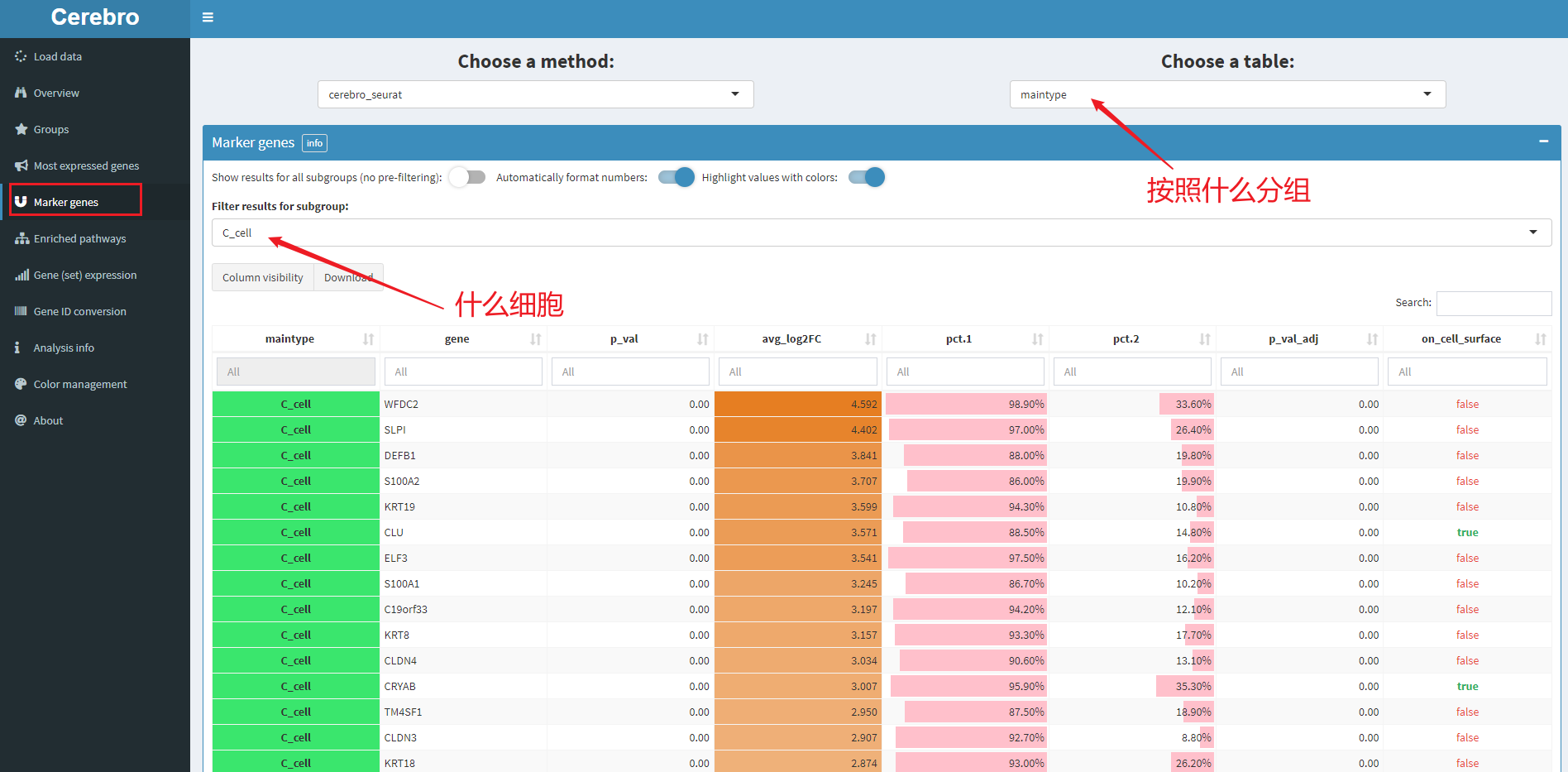
如图,表示C_cell中的差异基因。表格的展示也是很好看的,此外,这些表格都是可以下载的。
5.5 富集分析的结果展示
展示形式跟上面差不多,此处略过
5.6 基因表达、基因集评分的投影图

5.7 基因ID转换
以上是一些展示功能,基因转换这个小功能也很有用

本次分享就到这里。真心觉得是个不错的工具,不太擅长生信分析或者刚刚接触单细胞的朋友可以试试。















 482
482











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









