







RNA-seq 数据处理记录(2)
原始数据的处理
去除adapter
- 找到接头序列
可以通过建库的试剂盒在Illumina官网查找,也可以通过trim_galore自动找到接头并去除。
conda install trim-galore
trim_galore -q 25 --phred33 --stringency 3 --length 36 --paired *_1.fq.gz *_2.fq.gz --gzip -o ./cleandata/trim_galoredata/
参数解释:
–quality:设定Phred quality score阈值,默认为20,一般设为25.
–phred33::选择-phred33或者-phred64,表示测序平台使用的Phred quality score。具体怎么选择,看你用什么测序平台。
–adapter:输入adapter序列。也可以不输入,Trim Galore!会自动寻找可能性最高的平台对应的adapter。自动搜选的平台三个,也直接显式输入这三种平台,即–illumina、–nextera和–small_rna。
–stringency:设定可以忍受的前后adapter重叠的碱基数,默认为1(非常苛刻)。可以适度放宽,因为后一个adapter几乎不可能被测序仪读到。
–length:设定输出reads长度阈值,小于设定值会被抛弃。
–paired:对于双端测序结果,一对reads中,如果有一个被剔除,那么另一个会被同样抛弃,而不管是否达到标准。
–retain_unpaired:对于双端测序结果,一对reads中,如果一个read达到标准,但是对应的另一个要被抛弃,达到标准的read会被单独保存为一个文件。
–gzip和–dont_gzip:清洗后的数据zip打包或者不打包。
–output_dir:输入目录。需要提前建立目录,否则运行会报错。
– trim-n : 移除read一端的reads
代码示例:
trim_galore -q 25 --phred33 --stringency 3 --length 36 --paired CK-4_1.fq.gz CK-4_2.fq.gz --gzip -o ./cleandata/trim_galoredata/
去除random barcode
通过上面的步骤我们去除了原始序列中的adapter,但是在原始的read中还存在随机的random barcode或者是reads前面由于测序机器刚开始启动,会导致测序的碱基出现问题,这时候我们需要将其切除。
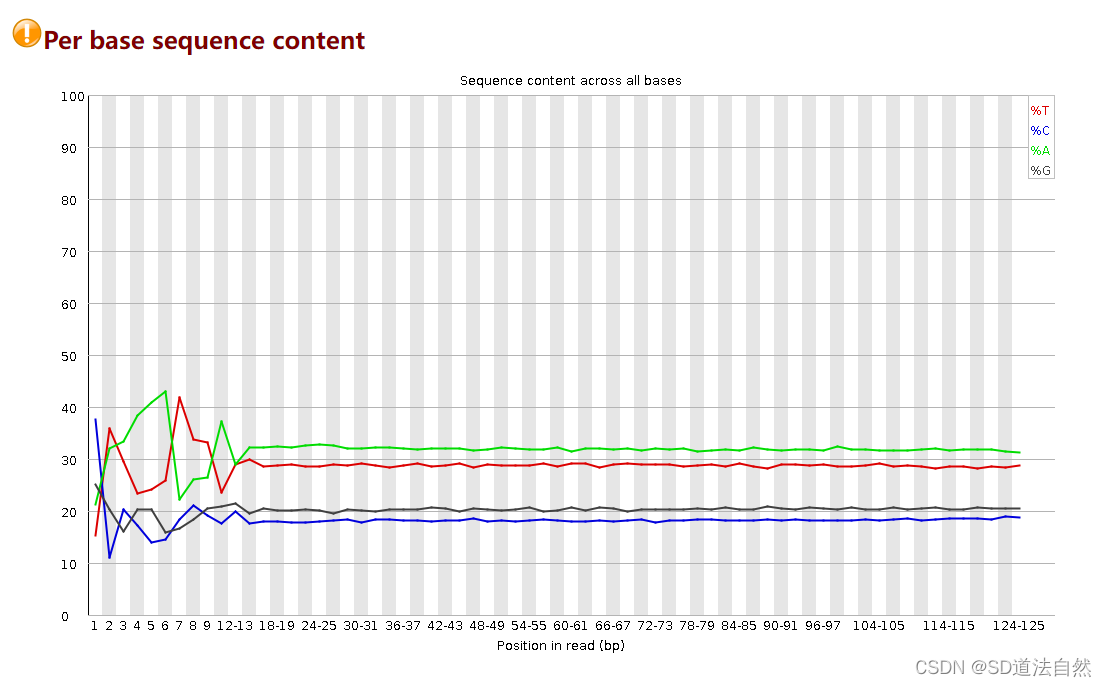
比如这里的前10个碱基
zcat xxx.fq.gz| fastx_trimmer -f 11 -l 125 -z -o ./cut_fastq/xxx.fq.gz
通过 fastx_trimmer 对前面的序列进行切割。(这里的序列虽然质量很高但是不是我们真正想要的序列而是加上去的,或者是机器的误读)
zcat 读取 压缩的文件
| 管道符,通过将zcat读取的文件传递给 fastx_trimmer
-f 截取序列的起始位置
-l 截取序列的末端
-z 为输出的文件是压缩形式的
-o 为输出的的文件名称
去除rRNA
- 原理
对于测序获得的原始的fastq由于接头序列的存在以及测序平台导致低质量的reads需要去除,这样才能保证最后的mapping质量。
目前有两种不同的建库方法,一种是直接通过polyA直接提取出mRNA,另一种建库方法是通过对rRNA进行去除(total RNA 中含量最多的就是rRNA),第二种方法得到的RNA其中包括了mRNA以及一些ncRNA,对于第二种建库的方法不一定能够保证对rRNA全部去除所以我们在建完库后仍然会有rRNA的存在这就会影响到我们下游的分析,所以我们需要在正式的mapping前,把reads中的rRNA去除。 - 操作
首先我们需要获取物种的rRNA,并建立索引,然后将原始的reads(已经去过接头的)比对到rRNA上,通过比对我们获取到没有比对上的reads,这样就可以获得去除rRNA的reads。
- 下载物种的rRNA序列
- 建立index
- mapping得到没有比对上的序列
下载物种的rRNA序列
ncbi

从NCBI中下载选择核酸数据库
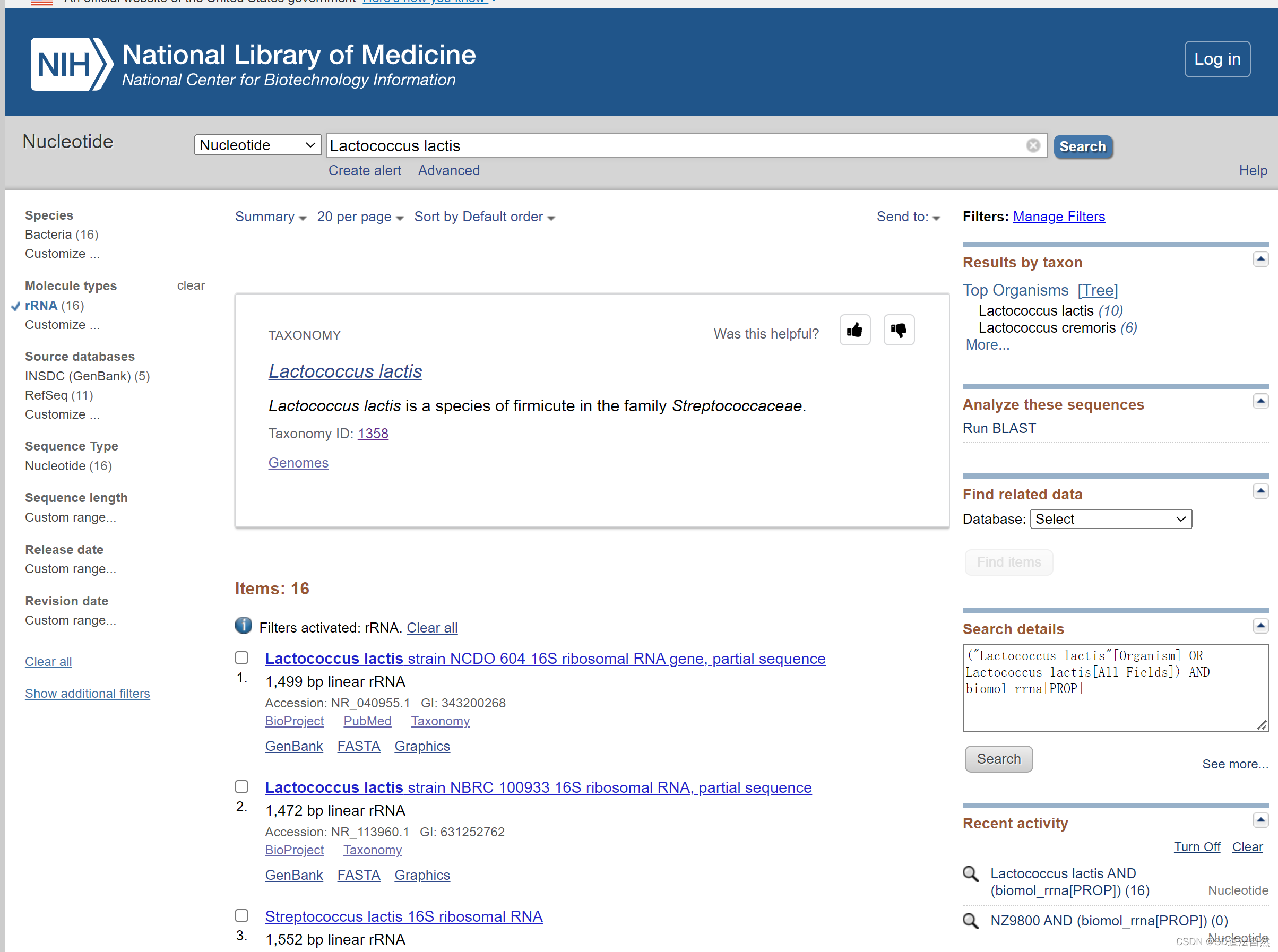
下载rRNA文件(原核生物:16SrRNA 23SrRNA 5SrRNA 真核生物: 18SrRNA)将其合并为一个fasta文件。
注:有时候NCBI中的16s 或者是18s可能不是很完整,所以需要通过另外一个数据库寻找完整的16s或者是18srRNA,silva通过这个数据库,可以找到完整的18srRNA以及16srRNA
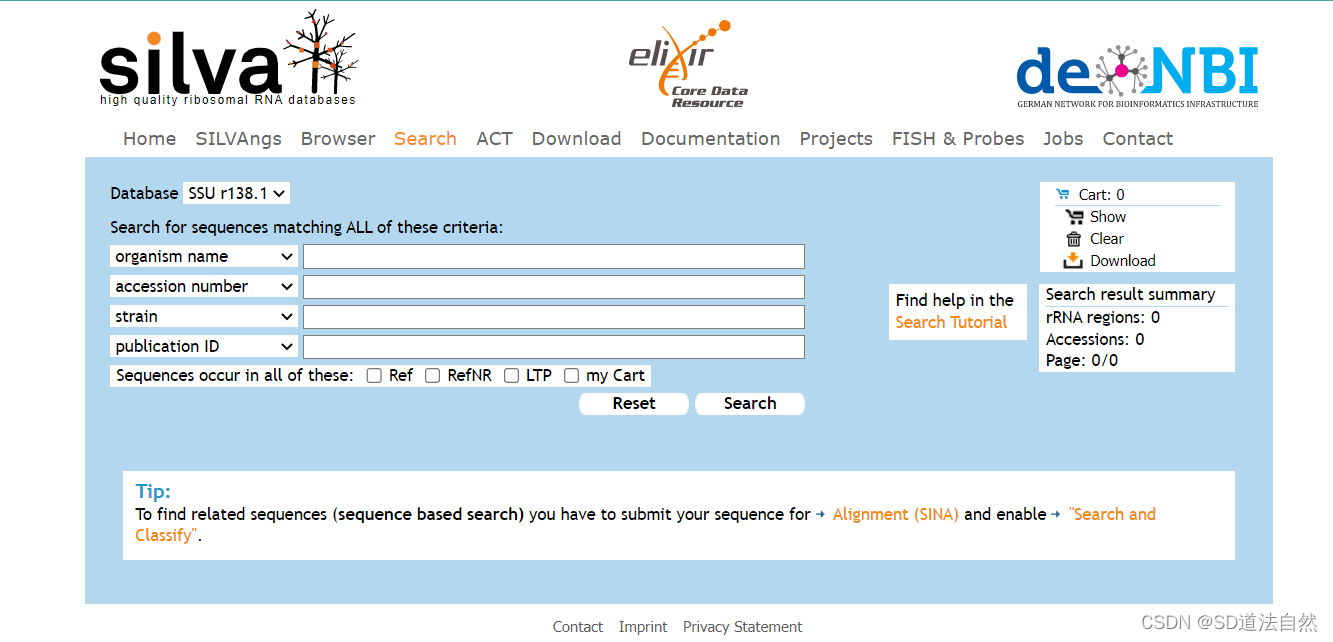
可以通过物种名进行搜索,在左上角可以看到是SSU(核糖体小亚基的数据库,16srRNA和18srRNA都是小亚基)搜索后就可以得到物种的16srRNA(原核生物),18srRNA(真核生物). 
选中下载
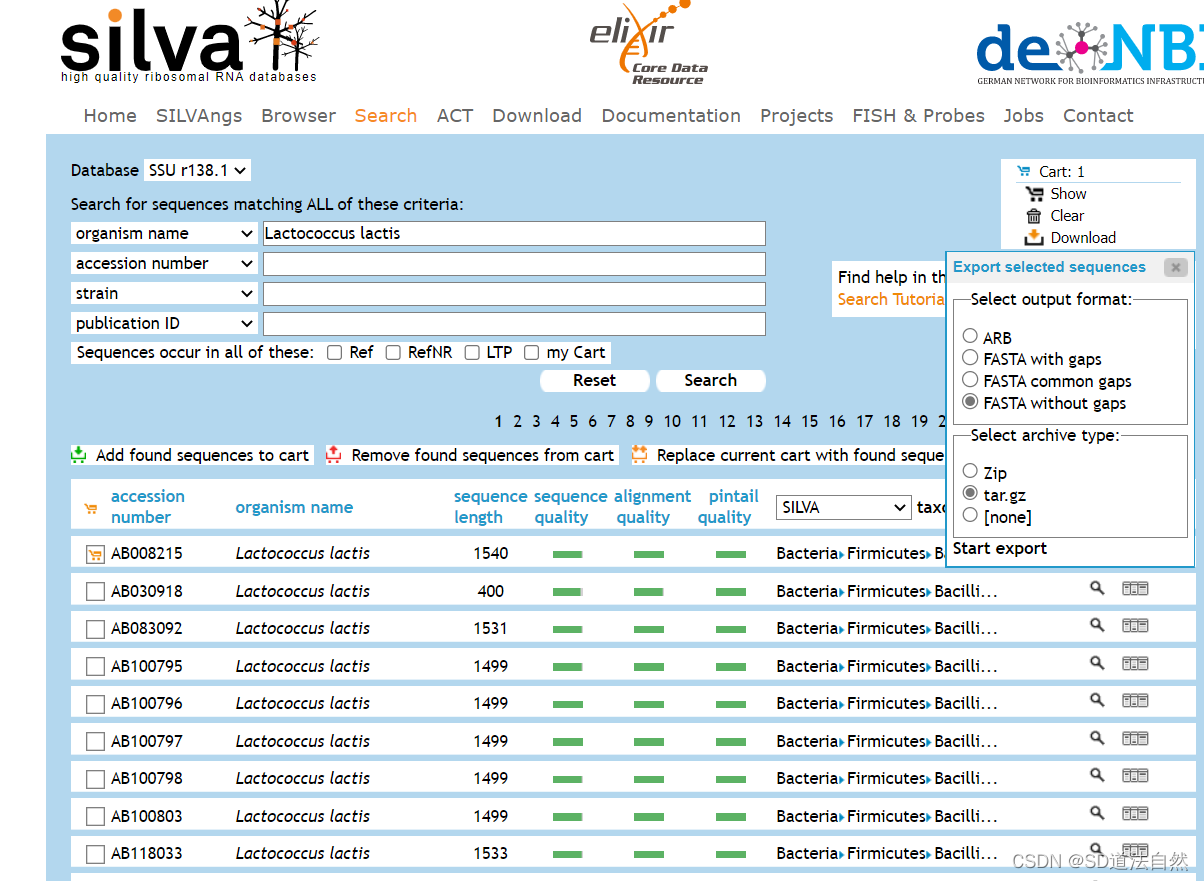
下载FASTA withoutgaps即可,点击start export
数据库会找寻一段时间状态显示为 Waiting

数据库状态显示为 finished 点击 Downloads file 下载即可
构建rRNA的index
构建rRNA的index使用bowtie2
conda install bowtie2
bowtie2-build rRNA.fa -p rRNA_index
首先使用conda 安装 bowtie2,然后使用bowtie2-build命令构建rRNA_index
参数:
rRNA.fa 为rRNA的序列
-p 为建立的index 命名,不加这个参数,会自动的将输入的fasta格式的文件作为index的名称。
rRNA_index 为 index的名字
去除原始数据中的rRNA_reads
bowtie2 -x ./rRNA/rRNA_index -1 ./clear_data/SRR8138826.man_1_val_1.fq.gz -2 ./clear_data/SRR8138826.man_2_val_2.fq.gz -p 16 -S ./sam_file/sam_out_bowie2.sam --un-conc-gz unmap_c_26
参数:
-x 指定bowtie2所需要的索引文件
-1 为双端测序文件中的reads1 (去除adapter)
-2 为双端测序文件中的reads2 (去除adapter)
-S 输入的sam文件名
–un-conc-gz 以压缩的形式输出没有比对上的文件(我们需要的去除rRNA的reads)
mapping 将处理过的reads比对到参考基因组上
这里我们使用的软件为tophat,tophat 是基于python2的,所以我们需要构建一个python2的环境,这里我们使用conda命令进行构建。
conda create -n py2env python=2.7
构建完环境后就可以开始比对了
首先构建索引:
bowtie2-build -f /public/Reference/GCF_020463755.1_ASM2046375v1_genomic.fa --threads 24 GCF_020463755.1_ASM2046375v1_genomic
-f 基因组序列文件
– threads 为调用的核心数
GCF_020463755.1_ASM2046375v1_genomic index的名称
tophat -o tophatout -G ./ref/ref.gtf -p 16 ./ref/GCF_020463755.1_ASM2046375v1_genomic unmap_c_24.1.gz unmap_c_24.2.gz
参数:
-o 为结果输出的文件夹
-G 需要的GTF格式的注释文件
- p 调用的核心数
./ref/GCF_020463755.1_ASM2046375v1_genomic bowtie2构建的index
unmap_c_24.1.gz read1(已经去除过adapt random barcode rRNA)
unmap_c_24.2.gz read2(已经去除过adapt random barcode rRNA)
使用cufflinks计算各个样本的FPKM
cufflinks -o {output} -p 16 -G {GTF} {input}
-o 输出目录
-p 核心数
-G 注释文件
{input} 上一步输出的SAM文件
cufflinks输出的结果:
(参考:
- transcripts.gtf
该文件包含Cufflinks的组装结果isoforms。前7列为标准的GTF格式,最后一列为attributes。其每一列的意义:
列数 列的名称 例子 描述
1 序列名 chrX 染色体或contig名;
2 来源 Cufflinks 产生该文件的程序名;
3 类型 exon 记录的类型,一般是transcript或exon;
4 起始 1 1-base的值;
5 结束 1000 结束位置;
6 得分 1000 ;
7 链 + Cufflinks猜测isoform来自参考序列的那一条链,一般是’+‘,’-‘或’.';
8 frame . Cufflinks不去预测起始或终止密码子框的位置;
9 attributes … 详见下
每一个GTF记录包含如下attributes:
Attribute 例子 描述
gene_idCUFF.1Cufflinks的gene id;
transcript_idCUFF.1.1 Cufflinks的转录子 id;
FPKM 101.267 isoform水平上的丰度,FragmentsPerKilobase of exon model perMillion mapped fragments; frac 0.7647 保留着的一项,忽略即可,以后可能会取消这个;conf_lo 0.07 isoform丰度的95%置信区间的下边界,即 下边界值 = FPKM * ( 1.0 - conf_lo );conf_hi 0.1102 isoform丰度的95%置信区间的上边界,即 上边界值 = FPKM * ( 1.0 + conf_hi ); cov 100.765 计算整个transcript上read的覆盖度;full_read_support yes 当使用 RABT assembly 时,该选项报告所有的introns和exons是否完全被reads所覆盖
- ispforms.fpkm_tracking
isoforms(可以理解为gene的各个外显子)的fpkm计算结果
- genes.fpkm_tracking
gene的fpkm计算结果Cuffmerge简介
Cuffmerge将各个Cufflinks生成的transcripts.gtf文件融合称为一个更加全面的transcripts注释结果文件merged.gtf。以利于用Cuffdiff来分析基因差异表达。















 2万+
2万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









