







scanpy和SingleCellExperiment和Seurat之间的转换
使用1
# 安装并加载 SeuratData中的pbmc3k数据集
# library(SeruatData)
# 1.---->install.packages("/Users/xiaokangyu/Desktop/单细胞学习/单细胞数据集/pbmc_3k/pbmc3k.SeuratData_3.1.4.tar.gz",repos=NULL,type="source")
# 2.---->library(pbmc3k.SeuratData)
# 3.---->pbmc3k=LoadData("pbmc3k")
rm(list=ls())
setwd("/Users/xiaokangyu/Desktop/单细胞学习/单细胞数据集/pbmc_3k/")
library(Seurat)
suppressMessages(library(SeuratDisk))
library(SeuratData)
## create Seurat object
#pbmc3k <- CreateSeuratObject(data,meta.data = label)
library(pbmc3k.SeuratData)
pbmc3k=LoadData("pbmc3k")
pbmc3k@meta.data$seurat_annotations=as.character(pbmc3k@meta.data$seurat_annotations)
SaveH5Seurat(pbmc3k, filename = "pbmc3k.h5Seurat")
Convert("pbmc3k.h5Seurat", dest = "h5ad")
scanpy转Seurat
```r
library(Seurat)
Convert_to_seurat3=function(adata){
mtx=py_to_r(adata$X$T$tocsc())
cellinfo=py_to_r(adata$obs)
cellinfo$cellname=rownames(cellinfo)
geneinfo=py_to_r(adata$var)
geneinfo$genename=make.unique(rownames(geneinfo))
colnames(mtx)=cellinfo$cellname
rownames(mtx)=rownames(geneinfo)
obj=CreateSeuratObject(mtx,meta.data = cellinfo[,!colnames(cellinfo)%in%c("n_genes","n_counts"),drop=F],min.features = 1)
return(obj)
}
library(reticulate)
use_condaenv("pytorch",required=T)
#py_config()
ad=import("anndata",convert = F)
adata=ad$read_h5ad("pbmc_ctrl_stim.h5ad")

使用注意3
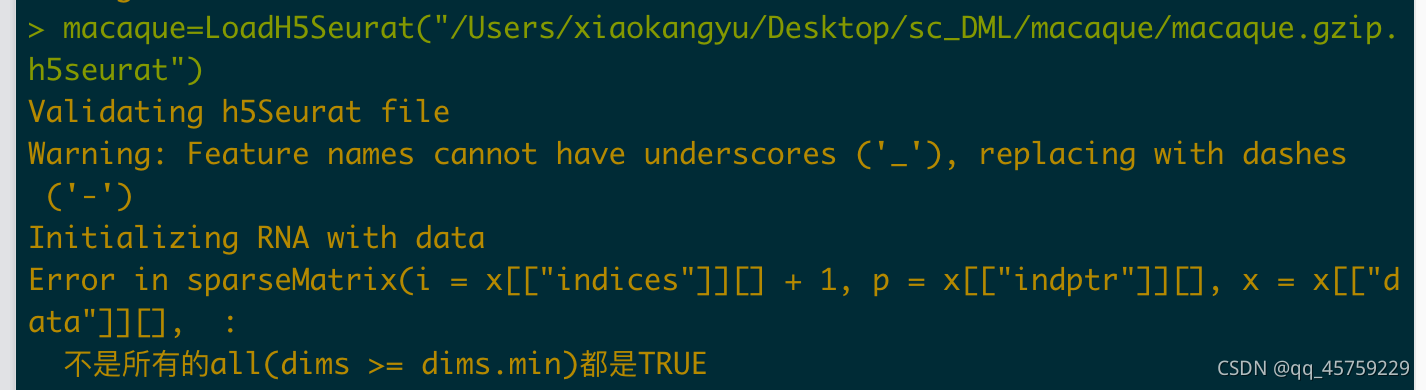 出现问题在于scanpy中的h5ad文件的adata.X的稀疏存储格式问题,需要转化成csr存储格式,正确的操作方式如下
出现问题在于scanpy中的h5ad文件的adata.X的稀疏存储格式问题,需要转化成csr存储格式,正确的操作方式如下
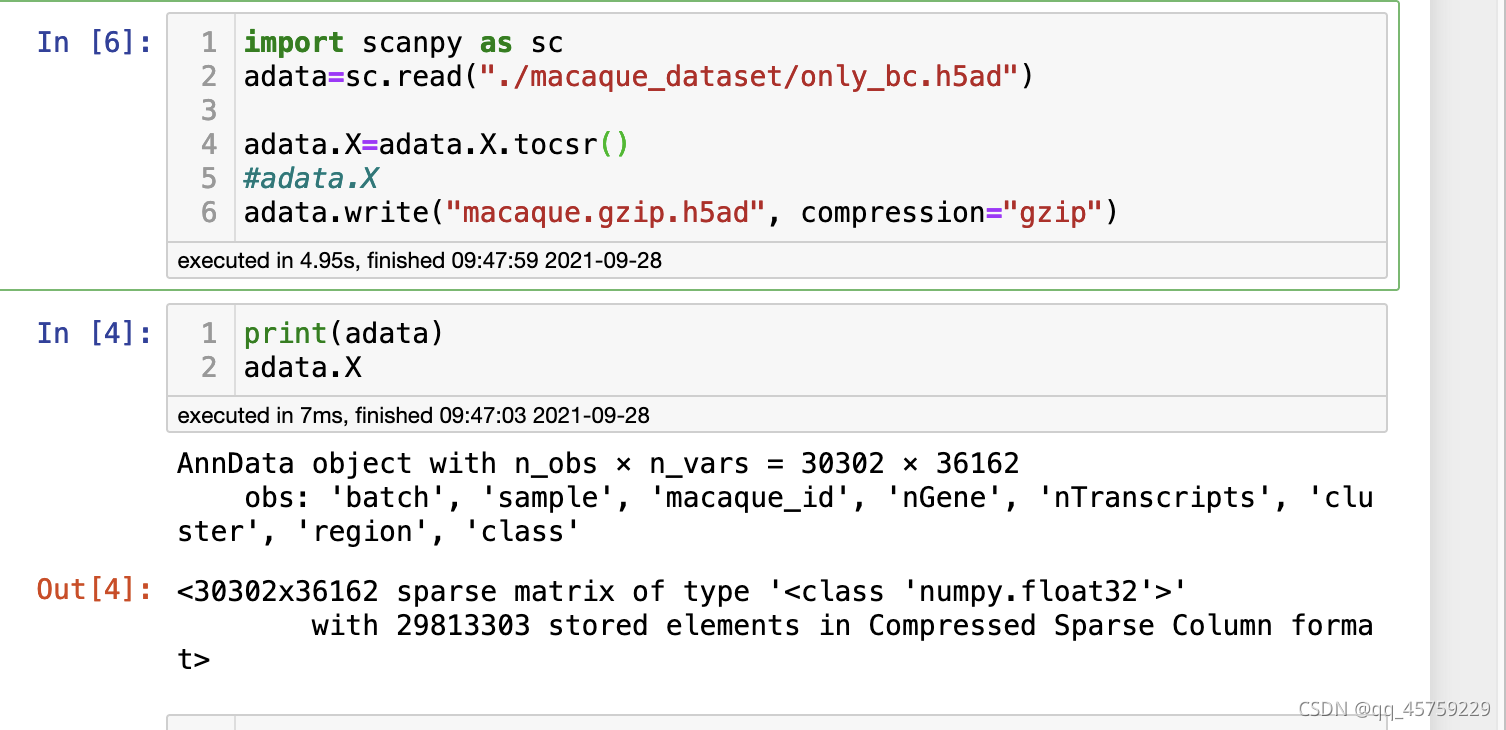 然后在R中使用
然后在R中使用
rm(list=ls())
## 方法1
library(Seurat)
setwd("~/Desktop/sc_DML/Bermuda/BERMUDA-master/macaque/")
Convert("/Users/xiaokangyu/Desktop/sc_DML/macaque/macaque.gzip.h5ad",dest = "h5seurat", overwrite = TRUE)
macaque=LoadH5Seurat("/Users/xiaokangyu/Desktop/sc_DML/macaque/macaque.gzip.h5seurat")
使用注意4
rm(list=ls())
suppressMessages(library(SingleCellExperiment))
suppressMessages(library(Seurat))
suppressMessages(library(SeuratDisk))
bct <- readRDS("SCE_MammaryEpithelial_x3.rds")
print(bct)#singlecellexperiment
# class: SingleCellExperiment
# dim: 1222 9288
# metadata(2): pumbed_ids marker_genes
# assays(2): counts logcounts
# rownames(1222): ENSMUSG00000092341
# ENSMUSG00000029580 ... ENSMUSG00000022949
# ENSMUSG00000028545
# rowData names(2): gene.symbols gene.counts
# colnames(9288): vis1 vis2 ... wal4374 wal4375
# colData names(4): study cell.class library_size
# detected_genes
# reducedDimNames(0):
# mainExpName: NULL
# altExpNames(0):
# 这里需要注意的是as.Seurat存在一个默认的参数data="logcounts",
# 之后会存在 aa@assays$originalexp里面
bct_seurat=as.Seurat(bct,data="counts")#也是没有问题的
# bct_seurat=as.Seurat(bct)会出现下面的错误
# Error in checkSlotAssignment(object, name, value) :
# assignment of an object of class “dgeMatrix” is not valid for slot ‘data’
# in an object of class “Assay”; is(value, "AnyMatrix") is not TRUE
# 需要加上 logcounts(bct)=as(logcounts(bct), "dgCMatrix")#必须加上这句,否则
# https://www.gormanalysis.com/blog/sparse-matrix-construction-and-use-in-r/
SaveH5Seurat(bct_seurat, filename = "bct_bct.h5Seurat")
Convert("bct_bct.h5Seurat", dest = "h5ad")
最终转换成的h5ad文件可以直接通过scanpy读取
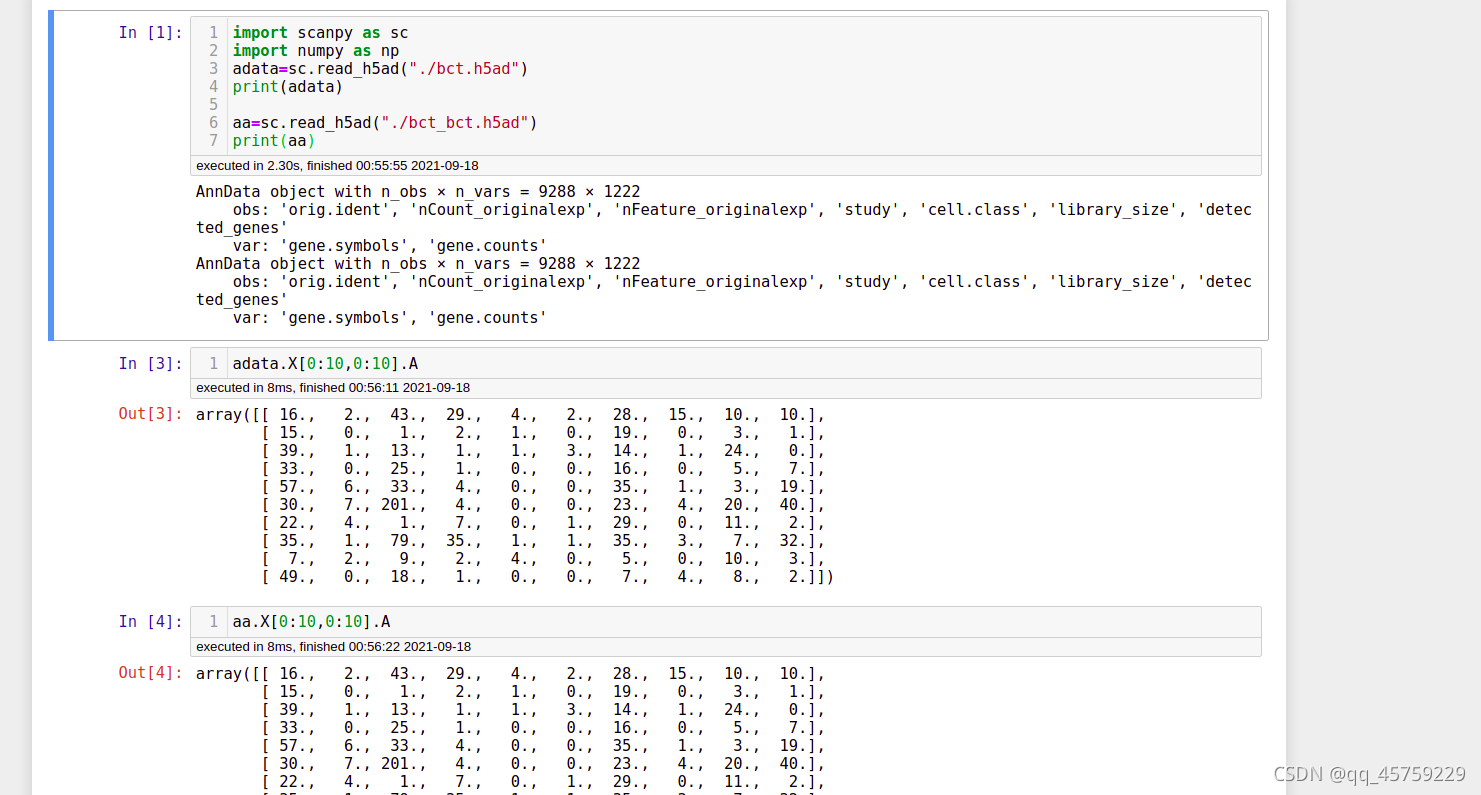
## 环境安装注意点
系统:macos
python:anaconda(base) python3.8.8
R: 4.1.1
当安装rpy2时,需要注意的是安装pip install rpy2==3.4.2版本,不要安装最新版本,否则下面的例子会出现问题
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-bF6bQGkY-1631809382770)(attachment:%E5%9B%BE%E7%89%87.png)]
```python
import scanpy as sc
import numpy as np
import anndata2ri
# Activate the anndata2ri conversion between SingleCellExperiment and AnnData
anndata2ri.activate()
#Loading the rpy2 extension enables cell magic to be used
#This runs R code in jupyter notebook cells
%load_ext rpy2.ipython
sc.settings.verbosity = 3
sc.logging.print_versions()
WARNING: If you miss a compact list, please try `print_header`!
The `sinfo` package has changed name and is now called `session_info` to become more discoverable and self-explanatory. The `sinfo` PyPI package will be kept around to avoid breaking old installs and you can downgrade to 0.3.2 if you want to use it without seeing this message. For the latest features and bug fixes, please install `session_info` instead. The usage and defaults also changed slightly, so please review the latest README at https://gitlab.com/joelostblom/session_info.
-----
anndata 0.7.6
scanpy 1.7.2
sinfo 0.3.4
-----
PIL 8.2.0
anndata2ri 1.0.6
anyio NA
appnope 0.1.2
attr 20.3.0
babel 2.9.0
backcall 0.2.0
backports NA
bottleneck 1.3.2
brotli NA
certifi 2020.12.05
cffi 1.14.5
chardet 4.0.0
cloudpickle 1.6.0
colorama 0.4.4
cycler 0.10.0
cython_runtime NA
cytoolz 0.11.0
dask 2021.04.0
dateutil 2.8.1
decorator 5.0.6
dunamai 1.6.0
fsspec 0.9.0
get_version 3.5
google NA
h5py 2.10.0
idna 2.10
ipykernel 5.3.4
ipython_genutils 0.2.0
jedi 0.17.2
jinja2 2.11.3
joblib 1.0.1
json5 NA
jsonschema 3.2.0
jupyter_server 1.4.1
jupyterlab_server 2.4.0
kiwisolver 1.3.1
legacy_api_wrap 1.2
llvmlite 0.36.0
markupsafe 1.1.1
matplotlib 3.3.4
mkl 2.3.0
mpl_toolkits NA
natsort 7.1.1
nbclassic NA
nbformat 5.1.3
numba 0.53.1
numexpr 2.7.3
numpy 1.20.1
packaging 20.9
pandas 1.2.4
parso 0.7.0
pexpect 4.8.0
pickleshare 0.7.5
pkg_resources NA
prometheus_client NA
prompt_toolkit 3.0.17
psutil 5.8.0
ptyprocess 0.7.0
pvectorc NA
pygments 2.8.1
pyparsing 2.4.7
pyrsistent NA
pytz 2021.1
requests 2.25.1
rpy2 3.4.2
scipy 1.6.2
send2trash NA
six 1.15.0
sklearn 0.24.1
sniffio 1.2.0
socks 1.7.1
sphinxcontrib NA
storemagic NA
tables 3.6.1
tblib 1.7.0
tlz 0.11.0
toolz 0.11.1
tornado 6.1
traitlets 5.0.5
typing_extensions NA
tzlocal NA
urllib3 1.26.4
wcwidth 0.2.5
yaml 5.4.1
zmq 20.0.0
zope NA
-----
IPython 7.22.0
jupyter_client 6.1.12
jupyter_core 4.7.1
jupyterlab 3.0.14
notebook 6.3.0
-----
Python 3.8.8 (default, Apr 13 2021, 12:59:45) [Clang 10.0.0 ]
macOS-10.15.7-x86_64-i386-64bit
8 logical CPU cores, i386
-----
Session information updated at 2021-09-16 23:35
%%R
suppressPackageStartupMessages(library(Seurat))
# Load PBMC dataset from Seurat tutorial
pbmc.data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19/")
# Initialize the Seurat object with the raw (non-normalized data).
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc
R[write to console]: Warning:
R[write to console]: Feature names cannot have underscores ('_'), replacing with dashes ('-')
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
Seurat->SingleCellExperiment->anndata
%%R -o pbmc_sce
#convert the Seurat object to a SingleCellExperiment object
pbmc_sce <- as.SingleCellExperiment(pbmc)
#convert singlecellexperment to Seurat
# as.Seurat()函数
pbmc_sce# R环境
class: SingleCellExperiment
dim: 13714 2700
metadata(0):
assays(2): X logcounts
rownames(13714): AL627309.1 AP006222.2 ... PNRC2.1 SRSF10.1
rowData names(0):
colnames(2700): AAACATACAACCAC-1 AAACATTGAGCTAC-1 ... TTTGCATGAGAGGC-1
TTTGCATGCCTCAC-1
colData names(4): orig.ident nCount_RNA nFeature_RNA ident
reducedDimNames(0):
mainExpName: NULL
altExpNames(0):
pbmc_sce# python环境中的pbmc_sce
AnnData object with n_obs × n_vars = 2700 × 13714
obs: 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'ident'
layers: 'logcounts'
show that the conversion worked
Seurat
%%R
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-5HV3MbyQ-1631809382772)(output_9_0.png)]
scanpy
mito_genes = pbmc_sce.var_names.str.startswith('MT-')
pbmc_sce.obs['percent_mito'] = np.sum(pbmc_sce[:, mito_genes].X, axis=1).A1 / np.sum(pbmc_sce.X, axis=1).A1
pbmc_sce.obs["nFeature_RNA"] = pbmc_sce.obs["nFeature_RNA"].astype(float)# 这里需要转换格式
sc.pl.violin(pbmc_sce, ['nFeature_RNA', 'nCount_RNA', 'percent_mito'], jitter=0.4, multi_panel=True)
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-ohVGtJNF-1631809382773)(output_11_0.png)]
anndata转化成singleCellExperiment
import scanpy.datasets as scd
adata_paul = scd.paul15()
WARNING: In Scanpy 0.*, this returned logarithmized data. Now it returns non-logarithmized data.
... storing 'paul15_clusters' as categorical
Trying to set attribute `.uns` of view, copying.
adata_paul
AnnData object with n_obs × n_vars = 2730 × 3451
obs: 'paul15_clusters'
uns: 'iroot'
%%R -i adata_paul
adata_paul # class: SingleCellExperiment ...
saveRDS(adata_paul,file="./paul.rds")# 将转化的singlecellexperiment以rds格式保存
最近研究了一下rds文件转h5ad的问题,我发现SeuratData那个库虽然可以成功转换,但是做的其实很垃圾,同样的rds文件12Mb,我用SeuratData转成h5ad文件,最终h5ad变成了84Mb,就离谱,但是我使用sceasy库进行的转换,最终的h5ad文件仅仅8Mb,虽然这个小的文件没有设么影响,我真正转换mouse brain的百万数据集时,原始的rds文件仅仅1.96Gb,SeuratData转成的h5ad直接变到了8Gb,而我经过sceasy转换后h5ad文件仅仅是1.69Gb,文件还变小了,之前SeuratData转换的h5ad直接导致我的内存爆炸,而且SeuratData还要产生一个同等大小的h5Seurat文件,简直太离谱了,下面记录一下使用sceasy转换的用法,这个真的是太好了
使用案例1(Seurat转h5ad)
library(sceasy)
library(reticulate)
library(Seurat)
library(SeuratData)
library(SeuratDisk)
use_condaenv('base')
file="pbmc3k_final.rds"
loompy=reticulate::import('loompy')
seurat_object=readRDS(file)## an old seurat: seurat2
# An old seurat object
# 13714 genes across 2638 samples
seurat_new=UpdateSeuratObject(seurat_object) ## a new seruat:Seurat3(4)
# An object of class Seurat
# 13714 features across 2638 samples within 1 assay
# Active assay: RNA (13714 features, 1838 variable features)
# 2 dimensional reductions calculated: pca, tsne
# sceasy::convertFormat(seurat_object, from="seurat", to="anndata",
# outFile='pbmc3k_final.h5ad')
sceasy::convertFormat(seurat_new, from="seurat", to="anndata",
outFile='pbmc3k_final_new.h5ad')
使用案例2(sce转h5ad)
# 使用案例1(Seurat转h5ad)
library(sceasy)
library(reticulate)
library(Seurat)
# library(SeuratData)
# library(SeuratDisk)
use_condaenv('base')
file="****.rds" # single cell experiment
loompy=reticulate::import('loompy')
sce_object=readRDS(file)## an old seurat: seurat2
sceasy::convertFormat(sce_object, from="sce", to="anndata",
outFile='****.h5ad')
可以和SeuratData对比
suppressMessages(library(SingleCellExperiment))
suppressMessages(library(Seurat))
suppressMessages(library(SeuratDisk))
bct <- readRDS("SCE_MammaryEpithelial_x3.rds")
print(bct)#singlecellexperiment
# 这里需要注意的是as.Seurat存在一个默认的参数data="logcounts",
# 之后会存在 aa@assays$originalexp里面
bct_seurat=as.Seurat(bct,data="counts")#也是没有问题的
# bct_seurat=as.Seurat(bct)会出现下面的错误
# Error in checkSlotAssignment(object, name, value) :
# assignment of an object of class “dgeMatrix” is not valid for slot ‘data’
# in an object of class “Assay”; is(value, "AnyMatrix") is not TRUE
# 需要加上 logcounts(bct)=as(logcounts(bct), "dgCMatrix")#必须加上这句,否则
# https://www.gormanalysis.com/blog/sparse-matrix-construction-and-use-in-r/
SaveH5Seurat(bct_seurat, filename = "bct_sce_to_h5ad_seurat.h5Seurat")
Convert("bct_sce_to_h5ad_seurat.h5Seurat", dest = "h5ad")
可以测试大小
Seurat3 转换成h5ad
我遇到了这么一个问题,就是说Seurat3的环境里(r3.6.3)无法安装SeuratDisk, 从而无法转换,但是可以采取首先将Seurat3存成rds格式,然后再在Seurat4的环境里(r4.1.2) upgrade到Seurat4,然后再转换
rm(list=ls())
library(Seurat)
library(SeuratData)
library(SeuratDisk)
print(packageVersion("Seurat"))
data=readRDS("~/Desktop/Test/seurat3.rds")
seurat_new=UpdateSeuratObject(data)
# sceasy::convertFormat(seurat_new, from="seurat", to="anndata",
# outFile='pbmc3k_final_new.h5ad')
SaveH5Seurat(seurat_new, filename = "~/Desktop/Test/aaa.h5Seurat",verbose = F,overwrite = T)
Convert("~/Desktop/Test/aaa.h5Seurat", dest = "h5ad",overwrite = T,verbose=F)
print("done")

















 2552
2552

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









