






Last Update: 17 March 2024 染山
参考脚本:
WGCNA Gene Correlation Network Analysis
https://mp.weixin.qq.com/s/5OUY5KDwgi05MlFrV_7Qjw
Network analysis with WGCNA
WGCNA R 包根据表达数据构建“加权基因共表达网络分析”。最初发表于 2008 年,引用如下:
-
Langfelder, P. and Horvath, S., 2008. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics, 9(1), p.559. -
Zhang, B. and Horvath, S., 2005. A general framework for weighted gene co-expression network analysis. Statistical applications in genetics and molecular biology, 4(1).
更多信息
-
Recent PubMed Papers -
Original WGCNA tutorials - Last updated 2016 -
Video: ISCB Workshop 2016 - Co-expression network analysis using RNA-Seq data (Keith Hughitt)
Installing WGCNA
install.packages(c("tidyverse", "magrittr", "WGCNA"))
Overview
The WGCNA pipeline is expecting an input matrix of RNA Sequence counts. Usually we need to rotate (transpose) the input data so rows = treatments and columns = gene probes.
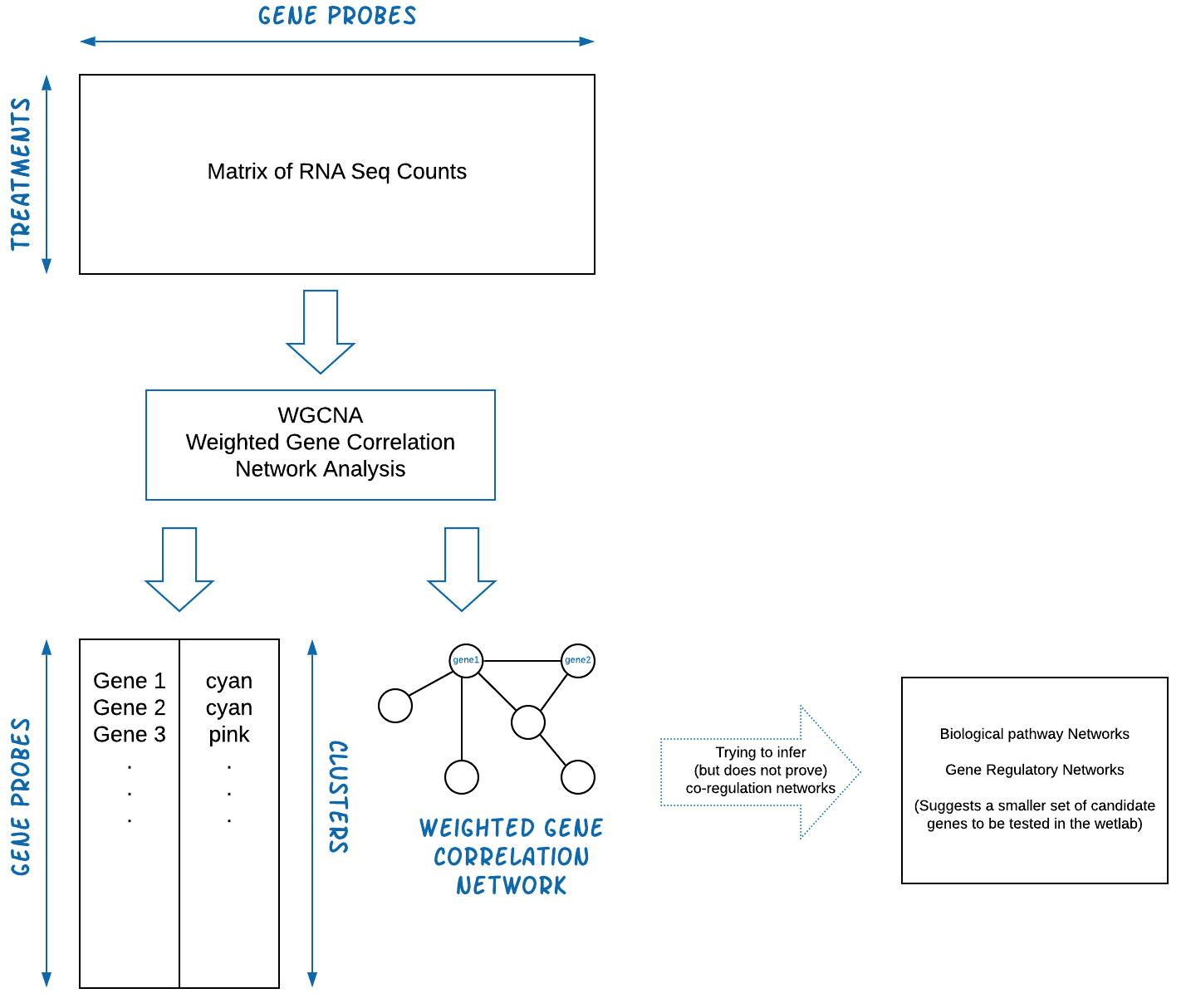
主要内容
WGCNA具体分析包含一下几个步骤
1、输入数据准备,样本筛选,该过程会产生聚类树图
2、计算软阈值为共表达网络建立基础该过程会产生Scale independence图和mean connectivity图
3、在上述基础上构建共表达网络,将基因划分到不同模块后,绘制基因聚类树 产生Cluster Dendrogram图
4、最重要一步绘制表型与基因相关性热图(WGCNA中最主要结果)产生Module-trait-relationships热图
5、探索感兴趣模块的基因与表型之间的相关性,输出图片为散点图
6、基因相关性热图
7、模块相关性热图
8、提取指定模块的基因做PPI网络分析
在 R 中安装WGCNA
install.packages("WGCNA") # WGCNA is available on CRAN
library(WGCNA)
WGCNA 在数据集较大时,普通电脑跑不动,需要借助服务器,在服务器中进行 WGCNA 分析时,我建议使用 Apptainer/Singularity 容器技术,避免在安装 R 包时各种系统依赖报错问题,如果您的服务器支持的话。在这里,作者自己通过 Apptainer 构建了一个 WGCNA 分析的容器 WGCNA.sif ,可以通过搜索微信公众号 Everything In Life 联系作者获取。

该容器已经安装了多个用于 WGCNA 分析的 R 包,可以直接使用,主要包括如下:
install.packages("BiocManager")
BiocManager::install("WGCNA")
BiocManager::install("tidyverse")
BiocManager::install("ggplot2")
BiocManager::install("ggrepel")
BiocManager::install("cowplot")
BiocManager::install("ggthemes")
install.packages("dplyr")
准备文件
-
样本表达量 expression.TPM.txt(至少5组数据 x 3 = 15 个样本)
$ head expression.TPM.txt
Gene A1 A2 A3 B1 B2 B3 C1 C2 C3 D1 D2 D3 E1 E2 E3
gene1 0.56 0 0 0 0.44 0 0 0 0 3.23 1.33 0 0 0 0
gene2 61.5 131.84 153.78 310.9 215.64 206.37 1.17 0.58 3.31 104.6 130.27 143.95 0.6 0.69 0.5
gene3 0.49 19.87 14.18 3.9 23.67 32.24 0.06 0.07 0 30.67 22.44 24.11 0 0.08 0
gene4 30.03 24.33 27 21.35 23.85 24.19 55.59 47.59 45.65 21.77 22.43 22.66 50.55 46.04 46
gene5 11 18.04 19.95 17.52 16.05 19.31 24.15 17.38 17.32 20.93 18.44 20.76 19.74 22.2 22
gene6 10.79 13.02 13.94 12.38 10.23 9.91 26.29 28.66 30.48 11.66 10.67 9.14 27.82 28.19 28
gene7 5.52 6.09 5.37 3.11 4.93 4.21 6.12 6.04 4.17 6.03 5.86 6.78 4.94 6.99 7
gene8 15.76 11.29 16.89 8.65 10.93 9.54 25.07 28.71 21.99 8.96 10.59 9.67 25.5 27.49 26
gene9 52.07 41.23 62.85 44.86 41.59 35.54 35.11 33.68 33.86 34.51 45.75 33.81 32.58 35.61 35
-
样本信息表 datTraits.txt
$ head datTraits.txt
Samples group sampleRep groupNo
A1 A 1 1
A2 A 2 1
A3 A 3 1
B1 B 1 2
B2 B 2 2
B3 B 3 2
C1 C 1 3
C2 C 2 3
C3 C 3 3
-
R 包运行代码 WGCNA.R
-
Apptainer 容器 WGCNA.sif(可选)
WGCNA.R 代码
################################################################################
# 1、数据准备,样本聚类,检查样本
# 准备好的数据进行样本聚类,聚类树查看,筛选查看是否存在异常样本,异常样本剔除
rm(list = ls()) #用于删除当前环境中的所有对象
library(WGCNA)
gene_exp <- read.csv(file = 'expression.TPM.txt',
sep = "\t",
row.names = 1)
# 去除第二列到最后一列之和等于0的行,即将表达量之和等于0的基因去除
WGCNA0 <- gene_exp[rowSums(data[2:ncol(data)]) != 0, ]
WGCNA0 <- gene_exp[rowSums(gene_exp[2:ncol(gene_exp)]) != 0, ]
#log2转换
WGCNA0 <- log2(WGCNA0+1)
#行列转置
WGCNA0 <- t(WGCNA0)
# # 先对基因进行筛选,var计算每个基因方差,筛选基因变化较大的基因,此次选取前50%的基因
# # 也可以不筛选基因全部基因去做
# vars_res <- apply(WGCNA0, 2, var)
# # 计算百分位数截止值
# per_res <- quantile(vars_res, probs = seq(0, 1, 0.25)) # quantile生成分位数
# per_res
#
# upperGene <- WGCNA0[, which(vars_res > per_res[3])] # 选取方差位于前50%的基因
# dim(upperGene)
#
# WGCNA1 <- data.matrix(upperGene)
#
# 如果已经有了关注的基因,则不建议对基因集进行过滤,因为可能会过滤掉你关注的基因。
# 不过滤无非就是多耗费的计算资源,在服务器上跑一般都没问题。
# 不使用方差筛选,直接使用所有基因
WGCNA1 <- data.matrix(WGCNA0)
nGenes <- ncol(WGCNA1)
nSamples <- nrow(WGCNA1)
head(WGCNA1[1:6,1:6])
library(stringr)
datTraits <- read.csv(file = 'datTraits.txt',
sep = "\t",
row.names = 1)
datTraits <- as.data.frame(datTraits)
head(datTraits)
#先检查
gsg <- goodSamplesGenes(WGCNA1, verbose = 3);
gsg[["allOK"]] # 全部合格
## [1] TRUE
# 如果是False则运行下面这部分
# if (!gsg$allOK){
# # Optionally, print the gene and sample names that were removed:
# if (sum(!gsg$goodGenes)>0)
# printFlush(paste("Removing genes:", paste(names(WGCNA1)[!gsg$goodGenes], collapse = ", ")));
# if (sum(!gsg$goodSamples)>0)
# printFlush(paste("Removing samples:", paste(rownames(WGCNA1)[!gsg$goodSamples], collapse = ", ")));
# # Remove the offending genes and samples from the data:
# WGCNA1 = WGCNA1[gsg$goodSamples, gsg$goodGenes]
# }
# 使用hclust函数对样本进行层次聚类,dist函数计算基因表达数据间的距离
sampleTree <- hclust(dist(WGCNA1), method = "average")
# 为每个样本的表型信息赋予不同的颜色,并生成颜色向量
traitColors <- numbers2colors(as.numeric(factor(datTraits$group)),
colors = rainbow(length(table(datTraits$group))),
signed = FALSE)
# 绘制样本树状图和表型热图,groupLabels参数用于添加表型标签
plotDendroAndColors(sampleTree,
traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")
# 根据样本树状图的可视化结果,手动选择合适的cutHeight值,将样本划分为不同的组
# clust <- cutreeStatic(sampleTree, cutHeight = 120, minSize = 10)
# 根据cutreeStatic函数的结果,将不符合要求的组或样本剔除,只保留符合要求的样本数据
# keepSamples <- (clust == 1)
# WGCNA1 <- WGCNA1[keepSamples, ]
# 计算剩余样本数据中的基因共表达网络,并获取基因数和样本数
# nGenes <- ncol(WGCNA1)
# nSamples <- nrow(WGCNA1)
# dim(WGCNA1)
save(WGCNA1, datTraits, nGenes, nSamples, file = "wgcna_step1.RData")
################################################################################
# 使用 load() 函数重新加载数据
load("wgcna_step1.RData")
library(WGCNA)
# 2、最优软阈值
# 数据准备样本聚类观测结束后,需要寻找最优软阈值(soft thresholding或power),
# 使构建的网络符合无标度拓扑结构。构建共表达网络
# 定义1:30的power值,用于构建不同的基因共表达网络
powers <- c(c(1:10),
seq(from = 12,
to = 30,
by = 2))
# 使用pickSoftThreshold函数,计算每个power值对应的scale free topology模型拟合程度和平均连接度,
# 并返回最佳的power值
sft <- pickSoftThreshold(as.matrix(WGCNA1),
powerVector = powers,
networkType = "signed")
# 根据sft对象中的结果,找到最佳的power值,并将其赋值给变量powerEstimate
powerEstimate = sft$powerEstimate
par(mfrow = c(1,2)) # 将绘图区域分为1行2列
cex1 = 0.9 # 定义文本大小
# 绘制scale independence随power变化的曲线
# 使用text函数添加标签,表示不同的power值
# 使用abline函数添加水平线,表示目标scale independence值(这里设置为0.9)
# Open a PDF device
pdf("power1.pdf", width = 5, height = 6) # Adjust width and height as needed
plot(sft$fitIndices[,1], # x轴为power值
-sign(sft$fitIndices[,3])*sft$fitIndices[,2], # y轴为scale independence指标
xlab = "Soft Threshold (power)", # x轴标签
ylab = "Scale Free Topology Model Fit,signed R^2", # y轴标签
type = "n", # 不绘制点和线
main = paste("Scale independence")) + # 图标题
text(sft$fitIndices[,1], # 在每个点上添加标签
-sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels = powers,
cex = cex1,
col = "steelblue") +
abline(h = 0.80, # 添加水平线
col = "red")
# Save the first plot to the PDF
dev.off() # Close the current PDF device
# 绘制mean connectivity随power变化的曲线
# 使用text函数添加标签,表示不同的power值
pdf("power2.pdf", width = 5, height = 6)
plot(sft$fitIndices[,1], # x轴为power值
sft$fitIndices[,5], # y轴为mean connectivity指标
xlab = "Soft Threshold (power)", # x轴标签
ylab = "Mean Connectivity", # y轴标签
type="n", # 不绘制点和线
main = paste("Mean connectivity")) + # 图标题
text(sft$fitIndices[,1], # 在每个点上添加标签
sft$fitIndices[,5],
labels = powers,
cex = cex1,
col = "steelblue")
dev.off()
## 输出文件解读
##
## Scale independence图
##
## 绘制power值对应的散点图;结果显示从1开始整个变化趋势都还很明显,
## 而某个值以后趋势已经不明显了;因此选取这数值作为最佳的power值。
## Power一般R方要高于0.8。如果调试后还不能满足可能是表达矩阵没有处理恰当,
## 或者不满足分析条件比如样本里太少,基因数目过多等原因
##
## mean connectivity图
##
## 该图为不同软阈值情况下的网络连通度,平均连接度和power取值间关系选取power,一般在平均连接度变化呈平稳处
save(sft , file = "wgcna_step2.RData")
################################################################################
# 使用 load() 函数重新加载数据
load("wgcna_step1.RData")
load("wgcna_step2.RData")
library(WGCNA)
# 3、构建共表达网络
# 基于最优软阈值构建共表达网络,将基因划分到不同模块后,可以绘制基因聚类树:
enableWGCNAThreads() # 启用WGCNA多线程加速
if(T){ # 使用blockwiseModules函数,将基因共表达网络划分为不同的模块,
#并将结果存储在net对象中
net = blockwiseModules(
as.matrix(WGCNA1), # 基因表达矩阵
# power = sft$powerEstimate, # power值
power = 8,
maxBlockSize = nGenes, # 最大块大小
TOMType = "unsigned", # TOM类型
minModuleSize = 30, # 最小模块大小
reassignThreshold = 0, # 重分配阈值
mergeCutHeight = 0.25, # 合并高度阈值
numericLabels = TRUE, # 数字标签
pamRespectsDendro = FALSE, # PAM是否考虑树状图结构
saveTOMs = F, # 是否保存TOM矩阵
verbose = 3 # 输出详细信息的级别
)
table(net$colors) # 显示不同模块的基因数目
}
##模块可视化,分类树与色块对应图
pdf("plotDendroAndColors.pdf", width = 10, height = 6)
if(T){ # 判断条件为真
# 将模块标签转换为颜色,以便绘图
moduleColors=labels2colors(net$colors)
table(moduleColors) # 显示不同颜色的基因数目
# 绘制分类树和模块颜色对应图
plotDendroAndColors(net$dendrograms[[1]], moduleColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
}
dev.off()
## 输出文件解读
##
## Cluster Dendrogram
##
## 聚类树状图,这个图可以分为两部分看:上半部分是基因的层次聚类树状图,下半部分是基因模块,
## 也就是网络模块。上下对应,可以看到距离较近的基因(聚类到同一条分支)被划分到了同一模块。
## 每个颜色代表一个模块,灰色代表里面基因不属于任何模块。
save(moduleColors, net, file="wgcna_step3.RData")
################################################################################
# 使用 load() 函数重新加载数据
load("wgcna_step1.RData")
load("wgcna_step2.RData")
load("wgcna_step3.RData")
# 4、表型与基因相关性热图
library(WGCNA) # 载入WGCNA包
library(forcats) # 载入forcats包
datTraits$group <- factor(datTraits$group) # 将datTraits$group转换为因子变量
levels(datTraits$group) # 显示因子变量的水平
pdf("Module-trait_relationships.pdf", width = 10, height = 8)
if(T){
nGenes = ncol(WGCNA1) # 基因数目
nSamples = nrow(WGCNA1) # 样本数目
design <- model.matrix(~0+datTraits$group) # 构建模型矩阵
colnames(design)= levels(datTraits$group) # 修改列名,获取分组信息
MES0 <- moduleEigengenes(WGCNA1,moduleColors)$eigengenes # 计算模块特征向量
MEs = orderMEs(MES0) # 对模块特征向量排序
moduleTraitCor <- cor(MEs,design,use = "p") # 计算模块特征向量与表型的相关系数矩阵
moduleTraitPvalue <- corPvalueStudent(moduleTraitCor,nSamples) # 计算相关系数矩阵的p值
textMatrix = paste(signif(moduleTraitCor,2),"\n(",
signif(moduleTraitPvalue,1),")",sep = "") # 构建绘图时用的文本矩阵
dim(textMatrix)=dim(moduleTraitCor) # 修改文本矩阵的维度,与相关系数矩阵相同
par(mar=c(6, 8.5, 3, 3)) # 设置绘图边距
labeledHeatmap(Matrix = moduleTraitCor, # 绘制带标签的热图
xLabels = colnames(design), # x轴标签
yLabels = names(MEs), # y轴标签
ySymbols = names(MEs), # y轴符号
colorLabels = FALSE, # 不显示颜色标签
colors = blueWhiteRed(50), # 颜色范围
textMatrix = textMatrix, # 显示文本矩阵
setStdMargins = FALSE, # 不设置标准边距
cex.text = 0.5, # 文本大小
zlim = c(-1,1), # 颜色映射范围
main = paste("Module-trait relationships")) # 绘图标题
}
dev.off()
save(MEs, file = "wgcna_step4.RData")
## 输出文件解读
##
## Module-trait relationships
##
## 上图中,最左侧的颜色块代表模块,最右侧的颜色条代表相关性范围。
## 中间部分的热图中,颜色越深相关性越高,红色表示正相关,蓝色表示负相关;
## 每个单元格中的数字表示相关性和显著性。一般,我们会按相关性的绝对值筛选最相关模块,即负相关模块也应该考虑在内。
## grey模块中包含了所有未参与聚类的基因,因此是无效模块,不应用于后续分析。
################################################################################
# 使用 load() 函数重新加载数据
load("wgcna_step1.RData")
load("wgcna_step2.RData")
load("wgcna_step3.RData")
load("wgcna_step4.RData")
library(WGCNA)
# 5、感兴趣性状的模块相关性分析
# 感兴趣性状的模块与表型之间相关性分析,输出图片为散点图
## 找到以下这一行,仅修改模块名称
## selectModule<-c("purple")
pdf("verboseScatterplot.pdf", width = 8, height = 8)
if(T){
modNames = substring(names(MEs), 3) # 提取模块名称
geneModuleMembership = as.data.frame(cor(WGCNA1, MEs,
use = "p",method = "spearman")) #计算基因与模块的相关系数矩阵
MMPvalue = as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nSamples)) # 计算相关系数矩阵的p值
names(geneModuleMembership) = paste("MM", modNames, sep="") # 修改列名
names(MMPvalue) = paste("p.MM", modNames, sep="") # 修改列名
geneTraitSignificance <- as.data.frame(cor(WGCNA1,as.matrix(datTraits$groupNo),use = "p")) # 计算基因与表型的相关系数矩阵
GSPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance),nSamples)) # 计算相关系数矩阵的p值
names(geneTraitSignificance)<- paste("GS.",names(datTraits$group),sep = "") # 修改列名
names(GSPvalue)<-paste("GS.",names(datTraits$group),sep = "") # 修改列名
selectModule<-c("purple") # 选择要绘制的模块(这里只选择了一个)
# selectModule <- modNames # 批量作图
par(mfrow=c(ceiling(length(selectModule)/2),2)) # 设置绘图区域,批量作图开始
for(module in selectModule){
column <- match(module,selectModule) # 找到当前模块在geneModuleMembership中的列号
print(module) # 输出当前处理的模块名称
moduleGenes <- moduleColors==module # 找到属于当前模块的基因
# 绘制散点图,横轴为基因在当前模块中的模块内连通性,纵轴为基因与表型的相关系数
verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]),
abs(geneTraitSignificance[moduleGenes, 1]),
xlab = paste("Module Membership in", module, "module"),
ylab = paste("Gene significance for", module, "module"),
main = paste("Module membership vs. gene significance\n"),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)
}
}
dev.off()
# 输出文件解读 针对关键模块和感兴趣的性状进一步挖掘,看看基因与模块的相关性(Module Membership, MM) 和
# 基因与性状的相关性(Gene Significance, GS)之间是否有某种关联。
################################################################################
# 使用 load() 函数重新加载数据
load("wgcna_step1.RData")
load("wgcna_step2.RData")
load("wgcna_step3.RData")
load("wgcna_step4.RData")
library(WGCNA)
# 6、基因相关性热图
# 探索所有基因或关注基因之间的相关性,用热图呈现,颜色越深,说明基因之间的相互作用越强。这个图片锦上添花吧,没有表型与基因相关性热图重要。
# 主要是可视化 TOM矩阵,WGCNA的标准配图
# 然后可视化不同 模块 的相关性 热图
# 不同模块的层次聚类图
# 还有模块诊断,主要是 intramodular connectivity
# pdf("TOMplot.pdf", width = 15, height = 15)
if(T){
# 提取 WGCNA1 数据的基因树
geneTree = net$dendrograms[[1]]
# 计算 TOM 相似度矩阵
TOM=TOMsimilarityFromExpr(WGCNA1,power=6)
# 将 TOM 相似度矩阵转换为距离矩阵
dissTOM=1-TOM
# 将距离矩阵取7次方,以突出较大的距离差异
plotTOM = dissTOM^7
# 将对角线上的值设为 NA,以避免在可视化时影响效果
diag(plotTOM)=NA
# 绘制 TOM 图
pdf("6_allgene_Network-heatmap.pdf",width = 100,height = 100)
TOMplot(plotTOM,geneTree,moduleColors,main="Network heapmap plot of all genes")
# 选取 nSelect 个基因进行可视化
nSelect =200
set.seed(10)
select=sample(nGenes,size = nSelect)
# 提取选定基因的 TOM 相似度矩阵
selectTOM = dissTOM[select,select]
# 对选定基因的 TOM 相似度矩阵进行聚类分析,生成基因树
selectTree = hclust(as.dist(selectTOM),method = "average")
# 提取选定基因的模块颜色信息
selectColors = moduleColors[select]
# 将选定基因的距离矩阵取7次方,以突出较大的距离差异
plotDiss=selectTOM^7
# 将对角线上的值设为 NA,以避免在可视化时影响效果
diag(plotDiss)=NA
# 绘制选定基因的 TOM 图
TOMplot(plotDiss,selectTree,selectColors,main="Network heapmap of selected gene")
}
# dev.off()
################################################################################
load("wgcna_step1.RData")
load("wgcna_step2.RData")
load("wgcna_step3.RData")
load("wgcna_step4.RData")
library(WGCNA)
# 7、模块相关性热图
# 探索各个模块之间的相关性,这个图片也是锦上添花,没有表型与基因相关性热图重要。
pdf("plotEigengeneNetworks.pdf", width = 10, height = 10)
if(T){
MEs=moduleEigengenes(WGCNA1,moduleColors)$eigengenes # 计算模块特征向量
MET = orderMEs(cbind(MEs,datTraits$groupNo)) # 对模块特征向量和表型变量进行排序
par(cex = 0.9) # 设置字体大小
#png("step6-Eigengene-dendrogram.png",width = 800,height = 600) # 打开png图像文件
plotEigengeneNetworks(MET, "", marDendro = c(0,4,1,2), marHeatmap = c(3,4,1,2), cex.lab = 0.8, xLabelsAngle
= 90,excludeGrey = FALSE) # 绘制模块特征向量的网络图
}
dev.off()
# 输出文件解读
# 模块之间的聚类树和相关性热图,探索模块之间的互作关系
################################################################################
# 8、提取指定模块的基因做PPI网络分析
####gene export for VisANT or cytoscape
## module="purple" # 选择要导出的模块
## load() 函数重新加载数据
load("wgcna_step1.RData")
load("wgcna_step2.RData")
load("wgcna_step3.RData")
load("wgcna_step4.RData")
load("wgcna_step6.RData")
library(WGCNA) # 载入WGCNA包
# 获取所有模块的唯一值
allModules <- unique(moduleColors)
# 循环遍历所有模块
for (module in allModules) {
# 判断条件为真
if (TRUE) {
# 计算 TOM 相似度矩阵
TOM=TOMsimilarityFromExpr(WGCNA1,power=14)
probes = colnames(WGCNA1) # 获取基因名称
inModule = (moduleColors == module) # 找到属于当前模块的基因
modProbes = probes[inModule] # 提取属于当前模块的基因名称
head(modProbes) # 显示基因名称前几行
modTOM = TOM[inModule, inModule] # 提取属于当前模块的基因之间的TOM值
dimnames(modTOM) = list(modProbes, modProbes) # 修改维度名称
### 这里只选了top1000的基因
nTop = 10000 # 设置要选择的基因数目
IMConn = softConnectivity(WGCNA1[, modProbes]) # 计算当前模块中基因之间的相似性
top = (rank(-IMConn) <= nTop) # 找到相似性排名前nTop的基因
filterTOM = modTOM[top, top] # 提取相似性排名前nTop的基因之间的TOM值
# for cytoscape
cyt = exportNetworkToCytoscape(filterTOM,
edgeFile = paste("CytoscapeInput-edges-", paste(module, collapse="-"), ".txt", sep=""),
nodeFile = paste("CytoscapeInput-nodes-", paste(module, collapse="-"), ".txt", sep=""),
weighted = TRUE,
threshold = 0.02,
nodeNames = modProbes[top],
nodeAttr = moduleColors[inModule][top]) # 导出Cytoscape可用的网络数据
}
}
说明:使用 save(WGCNA1, datTraits, nGenes, nSamples, file = "wgcna_step1.RData") 保存数据和使用 load("wgcna_step1.RData") 重新加载数据有利于在 R 中分步运行。
运行
$ apptainer exec WGCNA.sif Rscript WGCNA.R
OR
$ Rscript WGCNA.R
文:染山
本文由 mdnice 多平台发布














 2万+
2万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









