







一、数据准备
一共四个物种:油菜(Brassica napus)、白菜(Brassicarapa)、甘蓝(Brassica oleracea)以及拟南芥(Arabidopsis thaliana)的cds文件,蛋白pep文件和gff3文件,数据均来源于Ensembl网站(https://plants.ensembl.org/index.html)
实例:
pep:
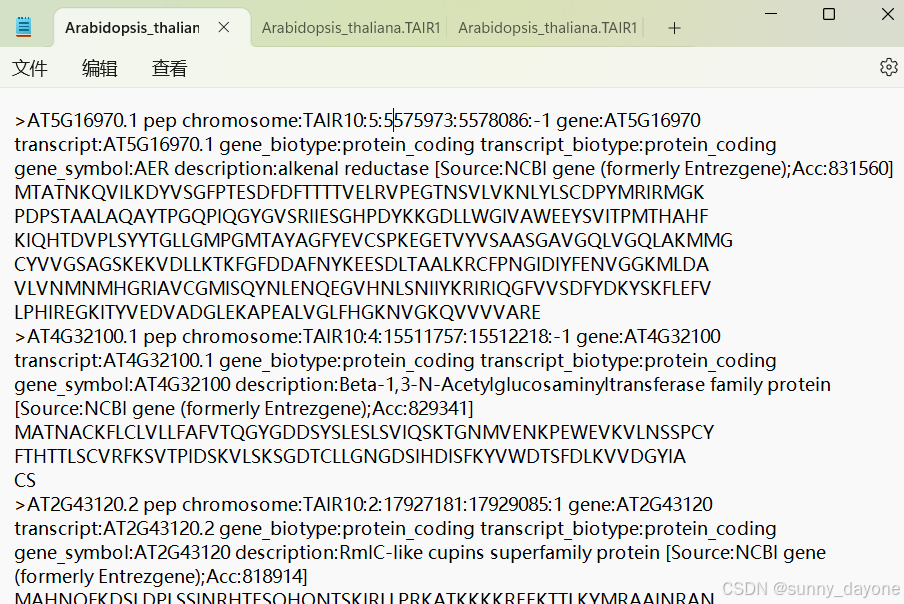
cds:
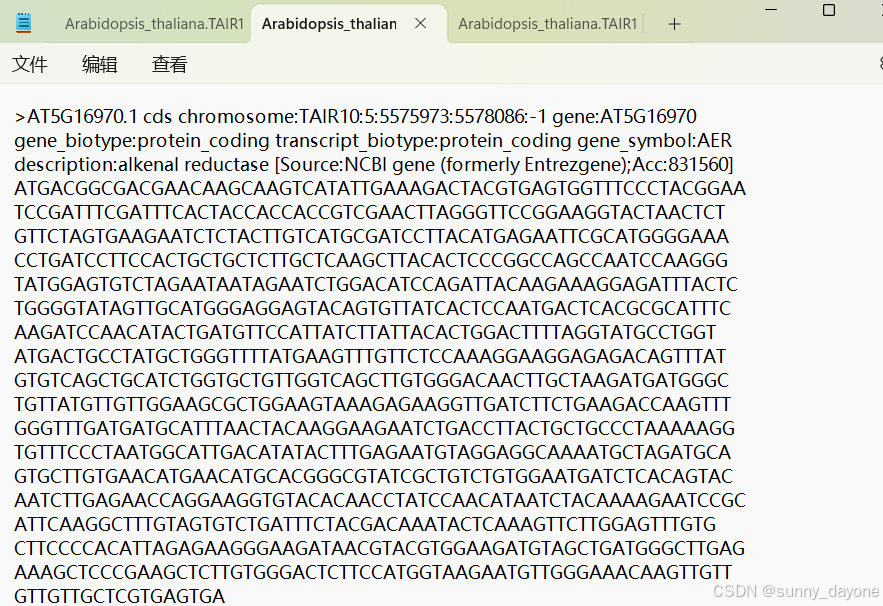
gff3
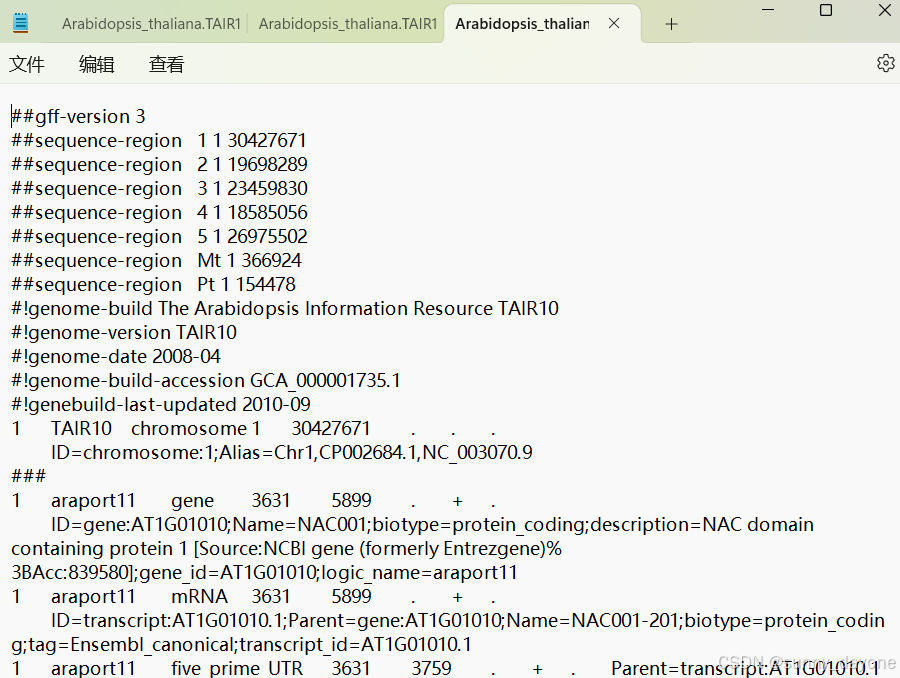
二、安装
##创建环境变量
conda create -n jcvi
##进入环境变量
codna activate jcvi
##安装jcvi
conda install -c bioconda jcvi
##安装依赖环境(除last外,其他非必须,可选)
conda install -c bioconda bedtools last ##bedtools last
sudo apt update && sudo apt upgrade
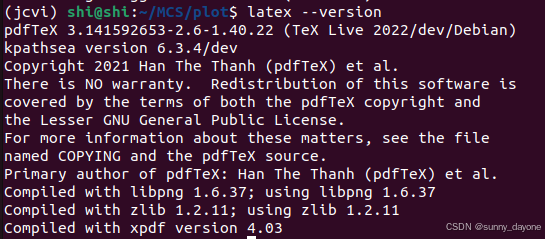
sudo apt install texlive-latex-extra -y ##LaTeX
latex --version ##确认安装版本
###############################
安装基本的TeXLive 包。如果你需要安装所有,可以使用 texlive-full,texlive-full 包含了所有的 TeX Live 包,这可能会占用大量的磁盘空间,会很慢很慢sudo apt-get install texlive-full
###############################
sudo apt-get install dvipng ##dvipng
- bedtools:用于处理基因组区间数据,有助于共线性分析的前处理和后处理步骤。
- last:用于全局和局部序列比对,可以提供用于共线性分析的比对结果。
- LaTeX 和 dvipng:用于生成高质量的报告和图形展示,但不是分析本身所必需的,但如果你需要发布或展示你的分析结果,它们可以帮助你创建专业和高质量的文档。
三、运行
(一)数据处理
1.将gff文件转换为bed文件
mkdir bed
python -m jcvi.formats.gff bed --type=mRNA ./gff3/Arabidopsis_thaliana.TAIR10.60.gff3 -o ./bed/At.bed
python -m jcvi.formats.gff bed --type=mRNA ./gff3/Brassica_napus.AST_PRJEB5043_v1.60.gff3 -o ./bed/Bn.bed
python -m jcvi.formats.gff bed --type=mRNA ./gff3/Brassica_oleracea.BOL.60.gff3 -o ./bed/Bo.bed
python -m jcvi.formats.gff bed --type=mRNA ./gff3/Brassica_rapa.Brapa_1.0.60.gff3 -o ./bed/Br.bed
######
--type=mRNA:
这个参数用于指定你想要从GFF3文件中提取的特定类型的特征,运行这个命令时,它会从GFF3文件中筛选出所有类型为 mRNA 的特征,并将它们转换为BED格式。也可指定其他类型,如gene、CDS、exion
--key(未在命令中显示,但通常与 --type 一起使用):
这个参数通常用于指定GFF3文件中用于识别特定特征的键值。默认为ID,在GFF3文件中,每个特征都有一个唯一的ID,其他键,如 Parent, Name, transcript_id等
如果你的数据来源于NCBI,要注意NCBI中mRNA、蛋白、基因组的序列ID不同,提取之后打开bed文件看一下第四列的ID,看和蛋白文件及cds文件(可以用TBtools提取)中的是否相同,若不同后续提不出来数据
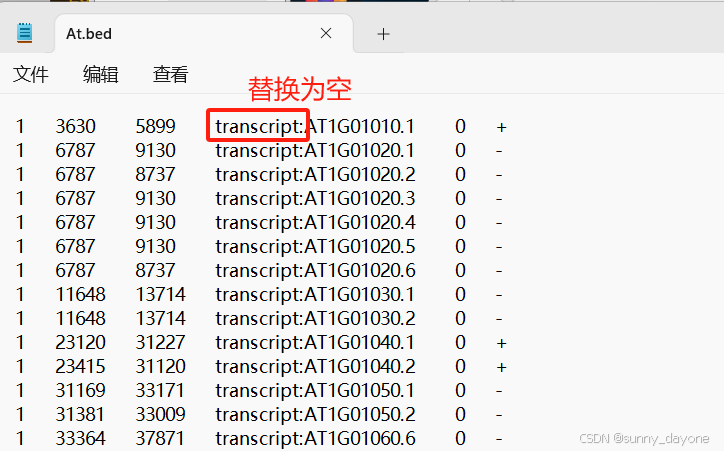
2.bed文件去重
主要目的是为了删除不同转录本,由于原始gff文件中是存在大量转录本信息,导致得到的bed文件存在大量位置重叠,因此需要取唯一值
python -m jcvi.formats.bed uniq bed/At.bed
python -m jcvi.formats.bed uniq bed/Bn.bed
python -m jcvi.formats.bed uniq bed/Bo.bed
python -m jcvi.formats.bed uniq bed/Br.bed
###########
得到X.uniq.bed文件和X.leftover.bed文件,我们只需要X.uniq.bed文件
X.uniq.bed: 这个文件包含的是去重后的bed 区域,即没有与其他区域重叠的独特区域。通常,工具会根据bed 文件中的染色体、起始位置和结束位置等信息来判断哪些区域是唯一的。
X.leftover.bed: 这个文件包含的是与其他区域有重叠的区域,或者可能是部分去重操作后剩余的区域。这些区域没有被认定为“唯一”或“独特”的
3.基于bed文件提取唯一转录本的CDS和pep
cut -f 4 bed/At.uniq.bed > bed/At.ids
cut -f 4 bed/Bn.uniq.bed > bed/Bn.ids
cut -f 4 bed/Bo.uniq.bed > bed/Bo.ids
cut -f 4 bed/Br.uniq.bed > bed/Br.ids
##cds
seqkit grep -f bed/At.ids cds/Arabidopsis_thaliana.TAIR10.cds.all.fa | seqkit seq -i > cds/At.cds
seqkit grep -f bed/Bn.ids cds/Brassica_napus.AST_PRJEB5043_v1.cds.all.fa | seqkit seq -i > cds/Bn.cds
seqkit grep -f bed/Bo.ids cds/Brassica_oleracea.BOL.cds.all.fa | seqkit seq -i > cds/Bo.cds
seqkit grep -f bed/Br.ids cds/Brassica_rapa.Brapa_1.0.cds.all.fa | seqkit seq -i > cds/Br.cds
###pep
seqkit grep -f bed/At.ids pep/Arabidopsis_thaliana.TAIR10.pep.all.fa | seqkit seq -i > pep/At.pep
seqkit grep -f bed/Bn.ids pep/Brassica_napus.AST_PRJEB5043_v1.pep.all.fa | seqkit seq -i > pep/Bn.pep
seqkit grep -f bed/Bo.ids pep/Brassica_oleracea.BOL.pep.all.fa | seqkit seq -i > pep/Bo.pep
seqkit grep -f bed/Br.ids pep/Brassica_rapa.Brapa_1.0.pep.all.fa | seqkit seq -i > pep/Br.pep注意:Brassica_rapa.Brapa_1.0.pep.all.fa 文件
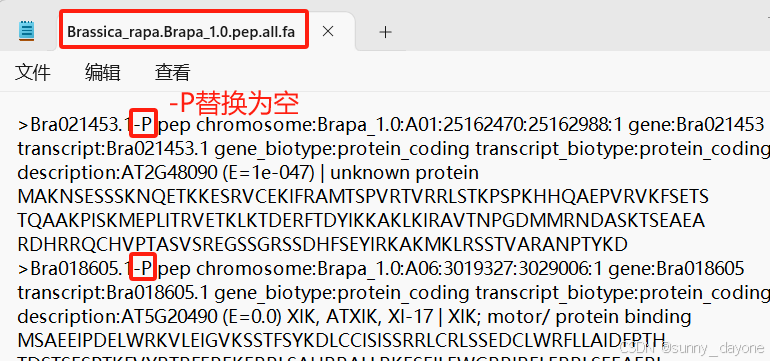
(二)jcvi 分析物种间的共线性区域
####创建新文件夹
mkdir -p plot && cd plot
cp ../cds/*.cds ./
cp ../pep/*.pep ./
cp ../bed/At.uniq.bed ./At.bed
cp ../bed/Bn.uniq.bed ./Bn.bed
cp ../bed/Br.uniq.bed ./Br.bed
cp ../bed/Bo.uniq.bed ./Bo.bed
python -m jcvi.compara.catalog ortholog --dbtype prot --no_strip_names At Br
python -m jcvi.compara.catalog ortholog --dbtype prot --no_strip_names At Bo
python -m jcvi.compara.catalog ortholog --dbtype prot --no_strip_names Bn Bo
python -m jcvi.compara.catalog ortholog --dbtype prot --no_strip_names Bn Br
python -m jcvi.compara.catalog ortholog --dbtype prot --no_strip_names Bn Bn
#######
主要研究油菜和其他三个物种之间的共线性关系,同时我们将油菜自己和自己进行比较,为后面将油菜分为A基因组和C基因组做准备
生成X.X.anchors文件、X.X.last文件、X.X.last.filtered文件、X.X.lifted.anchors文件和一些其他文件X.X.anchors:最终筛选出的共线性锚定点,代表可靠的相似基因对。用于共线性块的确定。
X.X.last::由 LAST 工具生成的原始比对结果,包含所有比对信息,包括大量冗余和重复匹配。.
作为进一步分析的基础数据。
X.X.last.filtered:进行初步筛选后的比对结果,去除了低质量比对。用于后续共线性分析中的锚定点提取。
X.X.lifted.anchors:对锚定点进行了抬升处理后的结果,以扩展共线性区域的覆盖范围。用于更加全面地识别共线性块。
若出现
LaTeX没安装好不用管,不影响出图
(三)可视化
需要bed文件、 X.X.anchors.simple文件、seqids文件和layout文件
1. X.X.anchors.simple文件
python -m jcvi.compara.synteny screen --minspan=30 --simple At.Br.anchors At.Br.anchors.new
python -m jcvi.compara.synteny screen --minspan=30 --simple At.Bo.anchors At.Bo.anchors.new
python -m jcvi.compara.synteny screen --minspan=30 --simple Bn.Bo.anchors Bn.Bo.anchors.new
python -m jcvi.compara.synteny screen --minspan=30 --simple Bn.Br.anchors Bn.Br.anchors.new
python -m jcvi.compara.synteny screen --minspan=30 --simple Bn.Bn.anchors Bn.Bn.anchors.new
#####
结果有两个文件X.X.anchors.new和X.X.anchors.simple。可视化需要X.X.anchors.simple文件
--minspan 设置 共线性块中基因的最小数量 ,过滤掉较小的、可能不太重要的对齐块,聚焦于较大的共线性块。
X.X.anchors.simple文件中的每一行都是一个共线性区块。前两列是一个物种开始和停止的基因,然后是另一个物种开始和停止的基因,最后两列是分数和方向,+为正向,-为反向。
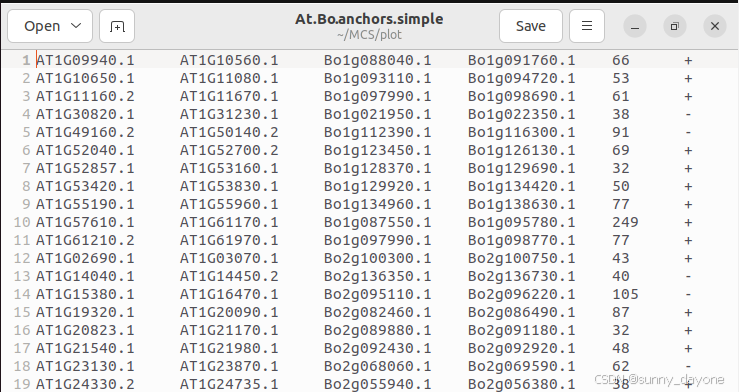
给特定共线性关系区域高亮的话,那么直接修改X.X.anchors.simple文件即可,只需要将想要高亮的那一行共线性关系区域的行首加你想改的颜色,颜色值为16进制,用“*”与ID隔开如#FF0000*AT1G10650.1,颜色的修改根据自己喜好修改即可。
2.seqids文件(需要自己准备)
seqids文件就是自己想要plot的染色体,下面是示例文件,染色体之间用英文逗号分隔(切记切记)。此文件的染色体编号名称需要和layout文件染色体编号名称对应,只是名称需要一致,顺序,数目可以不一致。
1,2,3,4,5
A01,A02,A03,A04,A05,A06,A07,A08,A09,A10
C9,C8,C7,C6,C5,C4,C3,C2,C1
LK031787,LK031789,LK031791,LK031793,LK031795,LK031797,LK031799,LK031801,LK031803
LK031803,LK031801,LK031799,LK031797,LK031795,LK031793,LK031791,LK031789,LK031787
3.layout文件(需要自己准备)
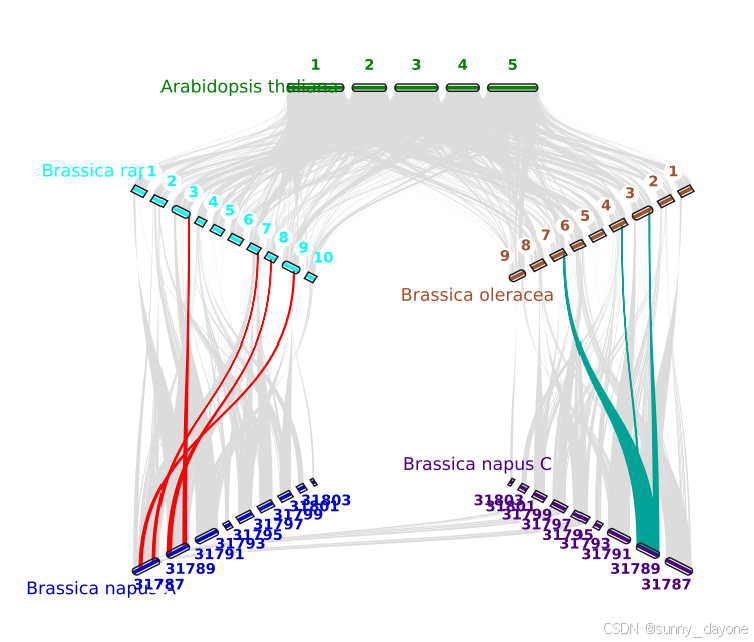
y, xstart,xend分别表示染色体在图上 Y 轴的位置(0-1),X 轴的起始和终止位置(0-1),rotation表示绘图时染色体倾斜的角度,color表示标签的颜色(颜色用英文或16进制表示都可以,也可以不填写即默认),label标签(物种名,可以根据自己的需要修改),va是染色体标签位置(top染色体标签在上,bottom在下),bed是物种的bed文件。
#edges 后是画共线性的数据文件,0,1,2,3,4分别代表第一二三四行的物种,
e, 0, 1, At.Br.anchors.simple 即进行Arabidopsis thaliana和Brassica rapa之间共线性的绘制
# y, xstart, xend, rotation, color, label, va, bed
.85, .4, .7, 0, , Arabidopsis thaliana, top, At.bed
.65, .2, .45, -30, , Brassica rapa, top, Br.bed
.65, .65, .9, 30, , Brassica oleracea, top,Bo.bed
.25, .2, .45, 30, , Brassica napus A, bottom, Bn.bed
.25, .65, .9, -30, , Brassica napus C, bottom, Bn.bed
# edges
e, 0, 1, At.Br.anchors.simple
e, 0, 2, At.Bo.anchors.simple
e, 1, 3, Bn.Br.anchors.simple
e, 2, 4, Bn.Bo.anchors.simple
e, 3, 4, Bn.Bn.anchors.simple
4.可视化
python -m jcvi.graphics.karyotype seqids layout
####报错Try running again with --notex option to disable latex.在使用 jcvi.graphics.karyotype 模块时,LaTeX 渲染出现问题
使用下方代码生成不使用LaTeX的图:
python -m jcvi.graphics.karyotype seqids layout --notex
###若想使染色体标签为染色体ID,使用下方代码
python -m jcvi.graphics.karyotype seqids layout --notex --keep-chrlabels报错findfont: Generic family 'sans-serif' not found because none of the following families were found: Helvetica意味着在系统上找不到默认的 sans-serif 字体系列没有管(LaTeX下不明白,崩溃了),能出图,除了丑没别的毛病
参考
共线性分析 | MCscan (Python version JCVI 包)
微信公众号:组学大讲堂《油菜花一样美!教你画漂亮的多物种基因家族共线性分析图!》
RPython 《基因家族分析流程08_物种间_共线性分析_kaks分析及可视化》
平凡生信人《物种间_共线性分析_kaks分析及可视化》

















 5154
5154

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









