









wgsim 就是这样一个软件,它是由开发了BWA等软件大牛 Li heng 写的基因组 转成 短序列的软件。
安装wgsim
~/app$ git clone https://github.com/lh3/wgsim.git && cd wgsim
~/app$ gcc -g -O2 -Wall -o wgsim wgsim.c -lz -lm
~/app$ sudo ln -s `pwd`/wgsim /usr/local/sbin
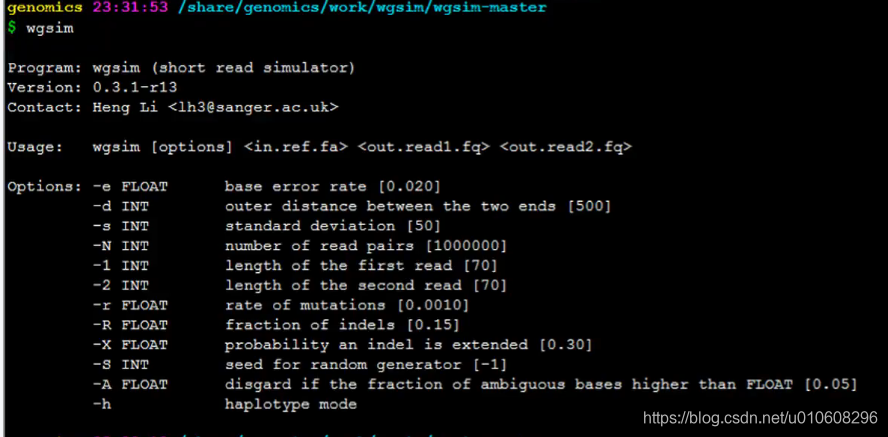
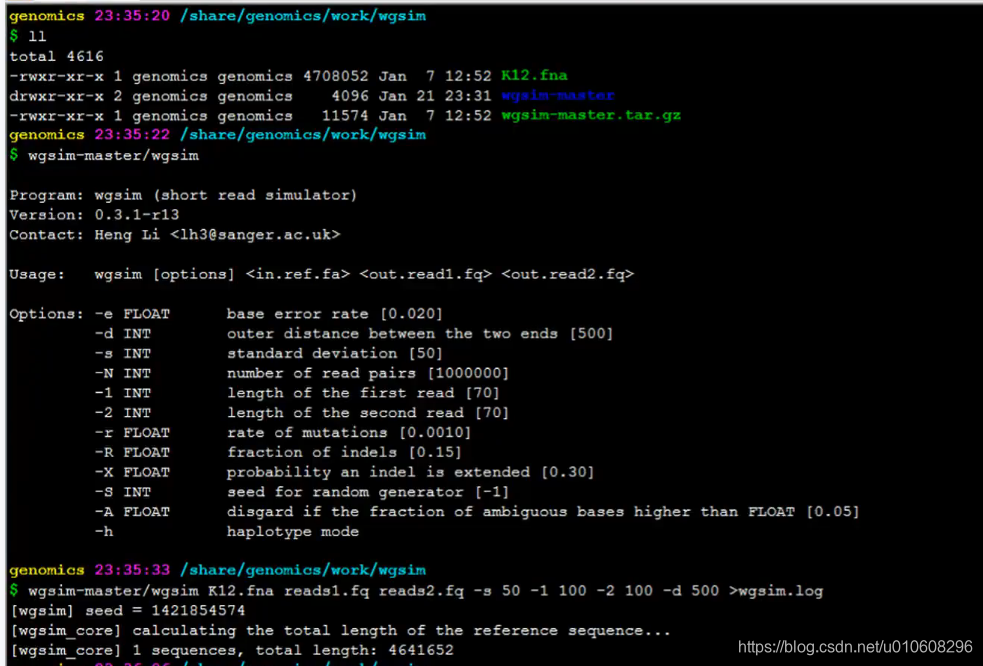
~/app$ wsgim -h
Program: wgsim (short read simulator)
Version: 0.3.1-r13
Contact: Heng Li <lh3@sanger.ac.uk>
Usage: wgsim [options] <in.ref.fa> <out.read1.fq> <out.read2.fq>
Options: -e FLOAT base error rate [0.020]
-d INT outer distance between the two ends [500]
-s INT standard deviation [50]
-N INT number of read pairs [1000000]
-1 INT length of the first read [70]
-2 INT length of the second read [70]
-r FLOAT rate of mutations [0.0010]
-R FLOAT fraction of indels [0.15]
-X FLOAT probability an indel is extended [0.30]
-S INT seed for random generator [-1]
-A FLOAT disgard if the fraction of ambiguous bases higher than FLOAT [0.05]
-h haplotype mode
模拟基因组短序列数据
使用所有参数为默认值,将大肠杆菌基因组数据转换为PE250 fastq 格式数据。
~/data$ wget https://raw.github.com/ecerami/samtools_primer/master/tutorial/genomes/NC_008253.fna
~/data$ wgsim -S11 -1250 -2250 NC_008253.fna reads_1.fastq reads_2.fastq
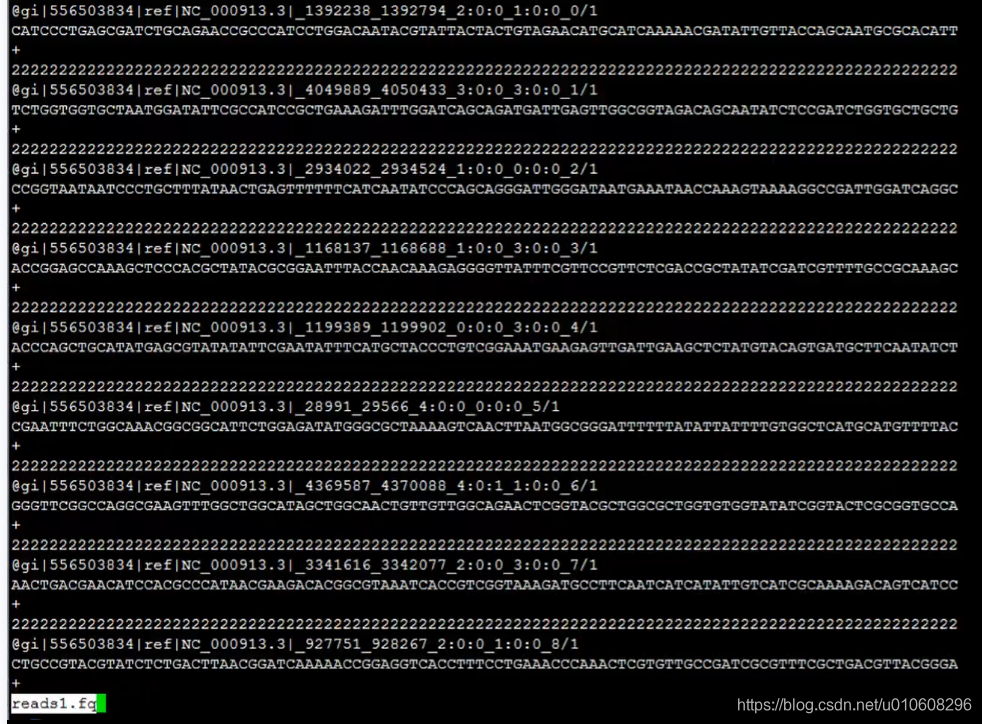
~/data$ head -8 reads_1.fastq
@gi|110640213|ref|NC_008253.1|_3151106_3151623_4:0:0_5:1:0_0/1
AACATAAGTGGTATTAATTCCCCAAGATTCAAGATTACGAATAGTATTATCCGCAAAAATATCATCACCTACTTTCGTCAGCATCAGGACTTTTGAATTCAATTTAGCCGCCGCCACCGCTTGATTAGCACCTTTGCCACCACATCCGATTTTGAAGGCAGGTGCTTCCAGAGTTTCTCCTTCTTTAGGCATCTGATAAGTGTAAGTAATGAGATCCACCATATTGGAACCAATAAGTGCAATGTCCATT
+
2222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222
@gi|110640213|ref|NC_008253.1|_2429297_2429873_8:0:0_7:0:0_1/1
ACGGTATCACCATGATTGCCCGTTCCGTCAACAGCATGGGGCTGGTCATTATAGGCGGTGGATCGCTTGAAGAAGCGTTAACTGAACTGGAAACCGGACGCGGCGACGCGGTGGTGGTGCTGGAAAACGAACTGCATCGTCACGCTTGCGCTACCCGCGTGAATGCTGCGCTGGCTAAAGCGCCGCTGGTGATTGTGGTTGACCATCAACGCACAGCGATTATGGAAAACGCTCATCTGGTACTATCCGC
+
222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222222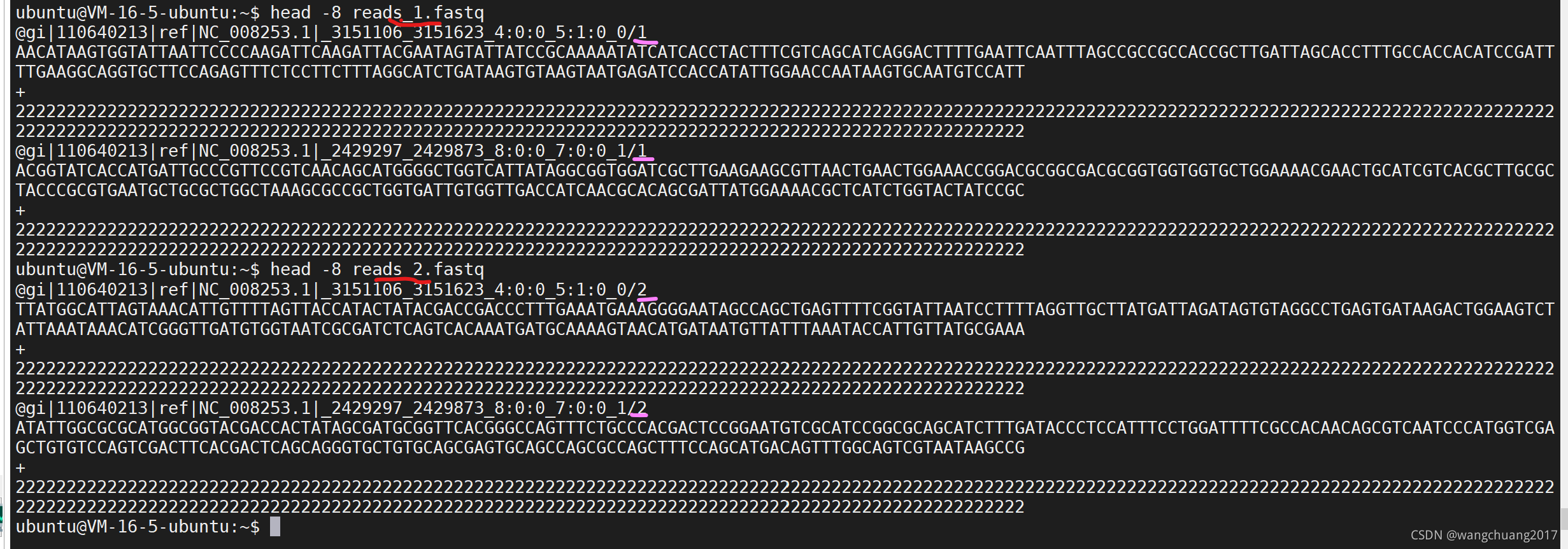


![]()


下载安装:
git clone https://github.com/lh3/wgsim.git
gcc -g -O2 -Wall -o wgsim wgsim.c -lz -lm
五、软件使用:
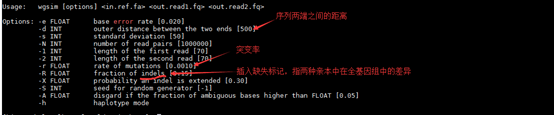
软件比较简单,输入文件为基因组序列,fasta格式,输出为illumina的fastq格式,这些格式我们在前面都介绍过。然后是一些选项。
-e 是错误率,默认是0.02
-d reads两头的距离,也就是插入片段长度,默认250bp,注意插入片段本身是包含reads长度的,而不是reads之间的距离
-s 是-d插入片段的偏差,默认是20,也就是-d的值加减20,我们知道插入片段长度并不是固定的,而是一个范围
-N 是测序的层数,控制输出数据量
-1 是reads1长度,默认70bp
-2 是reads2长度,默认70bp
-r 突变率
-R -X 都是调整indels的
-h 是单倍体模式
下面我们来运行一下
wgsim 参考序列 reads1 reads2 这里插入片段我们选择500bp,偏差-s在50,reads长度-1 -2为100bp,二者可以不一样,其余默认。
六、使用案例:
wgsim ref.fna reads1.fq reads2.fq -d 500 -s 50 -1 90 -2 90
七、注意事项:
1、模拟出的reads质量值是无法更改的,都是“I”,如果程序用到reads的质量值模拟数据就会有问题。
2、不支持Mate-pair文库,就是即使把-d设置微6K,那么它是不能像实际过程中发生环化的,两条reads的方向和小片段还是一样的。
介绍
============
Wgsim是从参照基因组中模拟序列的小工具。
它能够模拟二倍体基因组与SNP和插入/缺失(INDEL)
多态性,并能够模拟均匀替代测序错误的reads。
它不产生INDEL测序错误,但是这可能是部分地
通过模拟INDEL多态性补偿。
Wgsim输出是模拟多态性,并写入真正的reads坐标
以及在reads名称的多态性和测序错误的数量。
我们可以wgsim_eval.pl自带的包评估映射的准确性或SNP caller。
编译
===========
GCC -g -O2 -Wall -o wgsim wgsim.c -lz -lm
评估
==========
仿真与评估
-------------------------
仿真命令行:
wgsim -Nxxx -1yyy -d0 -S11 -e0 -rzzz hs37m.fa YYY-zzz.fq的/ dev / null的
其中yyy是read长,zzz为错误率和$ XXX * $ YYY =10000000。
默认情况下,多态性的15%以上是插入缺失和它们的长度是从绘制
几何分布密度0.7 * 0.3 ^ {L-1}。
评估命令行:
wgsim_eval.pl独特aln.sam | wgsim_eval.pl alneval -g 20
在'-g'选项可以和映射器来改变。
系统
------GCC: 4.1.2
CPU: AMD Opteron 8350 @ 2.0GHz
Mem: 128GB
实际操作:
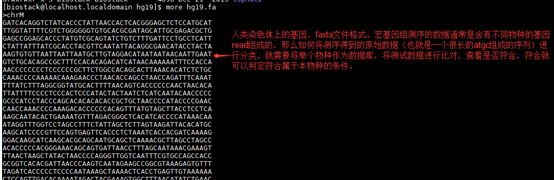
使用过的命令:
../seqtk_concat out_1.fq out_2.fq | head -n 8
../seqtk_concat out_1.fq out_2.fq > ../wgsim.fq #双端序列合并
seqtk_names wgsim.fq | cut -f1,2 -d'_'| cut -f3 -d'|' | perl -ane 's/chr\S+/9606/; print' | perl -ne 'print qq{$.\t$_}' | tabtk_bins - 1 >tabtk_stats
seqtk_names wgsim.fq | cut -f1,2 -d'_'| cut -f3 -d'|' | perl -ane 's/chr\S+/9606/; print' | perl -ne 'print qq{$.\t$_}' | tabtk_bins - 1 >tabtk_stats.tsv
cut -f1,2 tabtk_stats.tsv | tabtk_decorate /biostack/database/taxonomy/node-name.tsv 0 - > tab.xls
/*
解释:
perl -ane 's/chr\S+/9606/; print' 将每一行中包含chr并且其后非空格部分(比如chrab和chrbc)替换为9606
perl -ne 'print qq{$.\t$_}' 打印每一行的行号和内容;
可简化为:
|perl -ane 's/chr\S+/9606/; print qq{$.\t$_}' 或 perl -ane 's/chr\S+/9606/; print qq{$.\t$_}'
*/
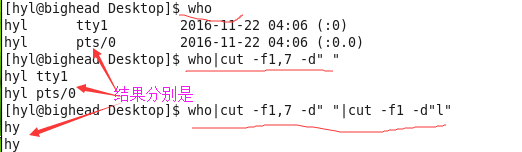
cut说明:
cut 不就是『切』吗?没错啦!这个指令可以将一段讯息的某一段给他『切』出来~ 处理的讯息是以
『行』为单位喔!
常用的参数:



















 2761
2761

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











