






通常而言测序的时候是不可能会测到5'接头,因为5'接头属于测序引物,我们测序是从5'到3', 整个插入片段测穿了也就是把3'接头测到而已。
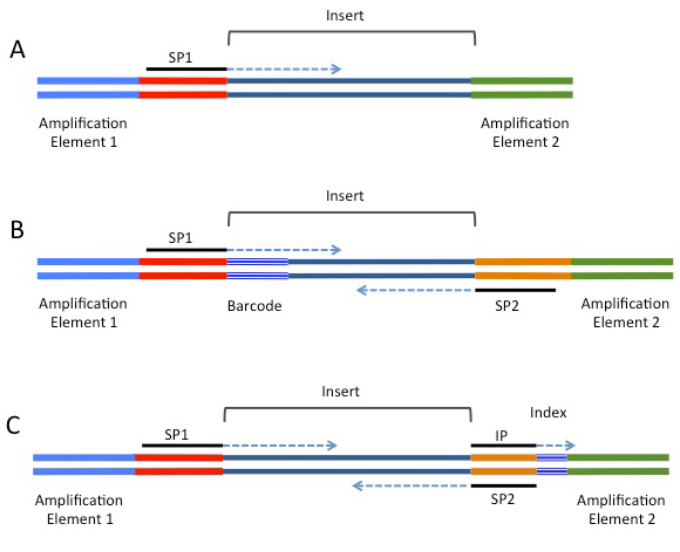
测序片段
但在建库过程中其实有一定概率会出现引物自连现象,也就是接头后面连着接头。当我们研究miRNA时,由于miRNA-seq长度只有22 nt左右,而测序P5接头序列长度也是这个范围,那么在胶回收的时候,这种短片段就会一起被回收,自然在后续测序时候也就可能会测到。这就是5' 接头污染现象。

文库构建流程
那么问题来了,这对我们数据分析有什么影响?
我以自己的一批数据作为一个例子,用cutadapt分析在原始序列中一共有多少条read是5'接头污染
RA5=GTTCAGAGTTCTACAGTCCGACGATC
cutadapt --max-n=0.1 -m 10 -g $RA5 --action=mask -o test.fq.gz test_raw.fq.gz
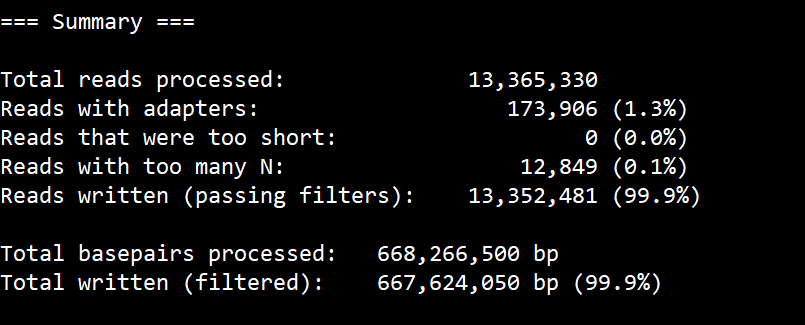
统计结果
仅有1.3%的原始数据是潜在的5'接头污染,也就是这批数据是合格的,不会影响我的分析。但是如果你的数据中存在大量的5'接头污染,你可以找公司聊聊了。
下一个问题就是如何处理5'污染的接头序列。一种方法,先过滤掉有5'接头的read,然后再用剩余的read进行3' 接头处理。另一种方法,是同时删除5' 和 3' 接头,然后按照长度过滤。
感觉上第一种方法更好些,只是要多写一步流程。我更偏爱第二种,因为高通量数据的量级摆在那里,只要数据整体没有问题,那么这些细节上的改变影响不了最后的结果。而如果你数据本身就很糟糕,那么别指望分析能给的数据带来显著的提高。
参考资料

















 568
568

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









