






 这篇教程介绍了如何利用MUMmer进行大片段序列比对,并通过R语言对比对结果进行可视化展示。首先,下载细菌基因组数据,使用nucmer进行比对并过滤出最佳匹配。接着,通过show-coords提取关键信息,并在R中读取这些信息。然后,绘制比对结果,不同颜色表示正向和反向匹配。最后,调整坐标轴并添加标签完成可视化。这种方法展示了基础的R语言绘图技巧,不同于ggplot2的图形语法。
这篇教程介绍了如何利用MUMmer进行大片段序列比对,并通过R语言对比对结果进行可视化展示。首先,下载细菌基因组数据,使用nucmer进行比对并过滤出最佳匹配。接着,通过show-coords提取关键信息,并在R中读取这些信息。然后,绘制比对结果,不同颜色表示正向和反向匹配。最后,调整坐标轴并添加标签完成可视化。这种方法展示了基础的R语言绘图技巧,不同于ggplot2的图形语法。
在2年之前我写过一篇教程介绍MUMmer软件的使用方法,可以通过如何使用MUMmer比对大片段序列阅读。
MUMmer作为一个比对工具,它的主要功能就是寻找两个序列的相似之处,至于如何展示结果,并不是它的主要目标。这篇文章将会介绍如何基于MUMMER的输出结果进行可视化。
首先是下载数据,我们用细菌的基因组作为案例
wget http://mummer.sourceforge.net/examples/data/H_pylori26695_Eslice.fasta
wget http://mummer.sourceforge.net/examples/data/H_pyloriJ99_Eslice.fasta然后使用nucmer进行比对
nucmer H_pylori26695_Eslice.fasta H_pyloriJ99_Eslice.fasta只保留1对1的最佳联配
delta-filter -1 out.delta > filter.delta输出容易展示的信息
show-coords -T -q -H filter.delta > coord.txt上面这几个程序的参数需要根据具体需求进行修改,并不是固定用法
后续就是在R语言里面继续绘图。 先加载数据并根据数据格式命名列
df <- read.table("~/coord.txt", sep = "\t")
colnames(df) <- c("ref_start", "ref_end", "qry_start", "qry_end", "ref_len", "qry_len",
"identiy", "ref_tag","qry_tag" )获取x和y轴的范围
x_range <- range(c(df$ref_start, df$ref_end))
y_range <- range(c(df$qry_start, df$qry_end))新建一个画图设备,并设置好画图区域
plot.new()
plot.window(xlim = x_range,
ylim = y_range)之后逐行绘制每个联配结果,用不同的颜色来展示倒置的情况
for( i in 1:nrow(df)){
if (df[i,3] < df[i,4]){
lines(x = df[i,1:2], y = df[i,3:4], col = "red")
} else{
lines(x = df[i,1:2], y = df[i,3:4], col = "blue")
}
}最后加上x轴和y轴的标签
box()
axis(1, at = seq(0, x_range[2], 10000), labels = seq(0, x_range[2], 10000) / 10000)
axis(2, at = seq(0, y_range[2], 10000), labels = seq(0, y_range[2], 10000) / 10000)结果如下
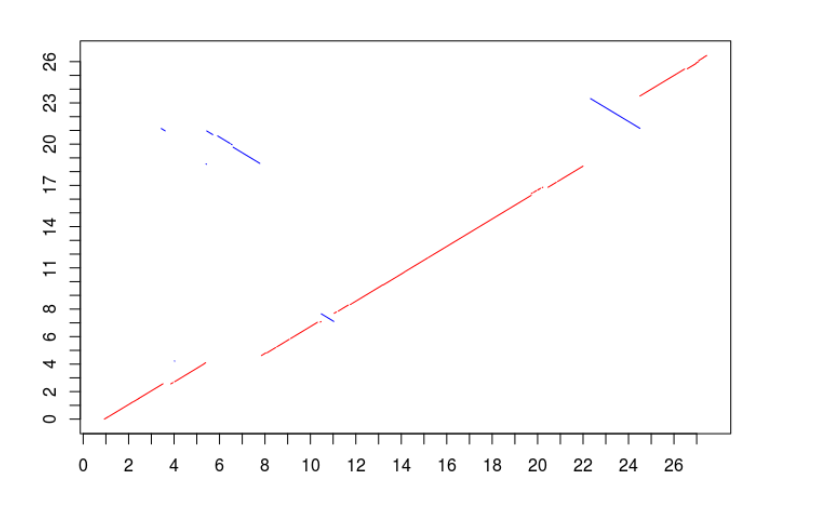
上面这种作图方式就是R语言的基础作图模式,从绘图思想上叫做画笔模式,这区别于ggplot2所代表的图形语法。

















 5888
5888

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









