






研究甲基化好久了,突然想整理一些常用分析方法,就先从差异甲基化筛选开始吧!
目标找到两组样本的甲基化差异位点,文件读入格式(此文件是从Bismark软件得到的结果并进行简单转化)如下:

首先按照R包:
install.packages(“methylSig_0.1.0.tar.gz”, repos=NULL, type=“source”)
调用此包:
library(methylSig)
读入文件包括4个急性淋巴血液病(AML)患者与4个正常人的甲基化位点:
fileList = c(system.file(“extdata”, “AML_1.txt”, package = “methylSig”), system.file(“extdata”, “AML_2.txt”, package = “methylSig”), system.file(“extdata”, “AML_3.txt”, package = “methylSig”), system.file(“extdata”, “AML_4.txt”, package = “methylSig”), system.file(“extdata”, “NBM_1.txt”, package = “methylSig”), system.file(“extdata”, “NBM_2.txt”, package = “methylSig”), system.file(“extdata”, “NBM_3.txt”, package = “methylSig”), system.file(“extdata”, “NBM_4.txt”, package = “methylSig”))
sample.id = c(“AML1”, “AML2”, “AML3”, “AML4”, “NBM1”, “NBM2”, “NBM3”, “NBM4”)
treatment = c(1,1,1,1,0,0,0,0)
#### Read Data ####
meth <- methylSigReadData(fileList, sample.ids = sample.id, assembly = “hg18”, treatment = treatment, context = “CpG”, destranded=TRUE)
甲基化差异位点分析:
myDiffSigboth = methylSigCalc(meth, groups=c(1,0), min.per.group=3)
可根据p值,q值定义差异甲基化位点:
myDiffSigbothDMCs = myDiffSigboth[myDiffSigboth[,“qvalue”] <= 0.05 & abs(myDiffSigboth[,“meth.diff”])>=25, ]
差异甲基化区域分析:
### Tiled analysis
methTile = methylSigTile(meth,win.size = 25)
myDiffSigbothTile = methylSigCalc(methTile, groups=c(1,0), min.per.group=3)
读到差异区域需要进行CpG岛注释:
library(“graphics”)
cpgInfo = getCpGInfo(system.file(“annotation”, “cpgi.hg18.bed.txt”, package = “methylSig”))
myDiffDMCs = myDiffSigboth[myDiffSigboth[,“qvalue”] < 0.05 & abs(myDiffSigboth[,“meth.diff”])>25,]
cpgAnn = cpgAnnotation(cpgInfo,myDiffSigboth)
cpgAnnDmc = cpgAnnotation(cpgInfo, myDiffDMCs)
cpgAnnotationPlot(cpgAnn,main=“ALL”)
cpgAnnotationPlot(cpgAnnDmc,main=“DMCs”)
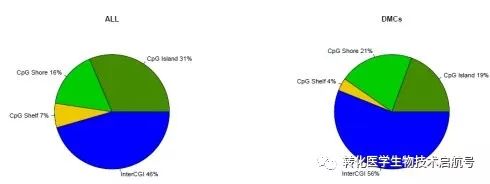
读到差异区域需要进行基因区域的注释:
refGeneInfo = getRefgeneInfo(system.file(“annotation”, “refGene.txt”,package = “methylSig”))
refGeneAnn = refGeneAnnotation(refGeneInfo, myDiffSigboth)
refGeneAnnDmc = refGeneAnnotation(refGeneInfo, myDiffDMCs)
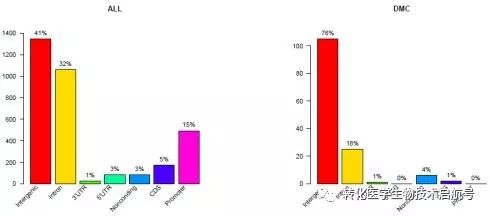
refGeneAnnotationPlot(refGeneAnn,main=“ALL”,priority=c(“promoter”,“cds”,“noncoding”, “5’utr”, “3’utr”))
refGeneAnnotationPlot(refGeneAnnDmc,main=“DMC”,priority=c(“promoter”,“cds”, “noncoding”, “5’utr”, “3’utr”))
6
关注公众号,每日有更新!
















 1314
1314











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









