







1.1 文章基本信息
- 标题
: Crowd-sourced benchmarking of single-sample tumor subclonal reconstruction
- 作者
: 多机构联合团队(包括Quentin Fottrell等)
- 期刊
: Nature Biotechnology
- 发表时间
: 2023年
- DOI
: 10.1038/s41587-023-01747-2
- 研究领域
: 肿瘤基因组学、计算生物学、亚克隆重建算法评估
1.2 文章摘要
本研究通过众包模式,系统评估了31种单样本肿瘤亚克隆重建算法的性能。通过生成高保真模拟肿瘤数据(涵盖不同突变负荷、克隆结构、测序深度等变量),研究团队量化了算法在检测亚克隆数目、细胞比例估计和突变聚类等任务中的准确性。结果表明,算法性能差异显著,部分算法(如PhyloWGS、PyClone-VI)在特定场景下表现优异,但整体仍存在高假阳性率、对低纯度样本敏感等局限。研究为算法选择和改进提供了数据支持,并开源了标准化评估框架。 —
—
1.3 研究背景与科学问题
- 背景
:
肿瘤异质性是癌症治疗耐药和复发的关键因素,单样本亚克隆重建通过解析肿瘤内不同亚群的突变和拷贝数变异(CNV),为精准治疗提供依据。 - 科学问题
:
现有算法在数据兼容性、准确性评估和临床适用性方面缺乏统一标准,导致临床转化困难。 - 核心问题
:
不同算法在复杂肿瘤场景下的鲁棒性如何?
哪些因素(如测序深度、肿瘤纯度)对算法性能影响最大?
1.4 研究方法与技术路线
- 数据生成
:
使用SERGIO模拟器生成包含不同克隆结构、突变负荷、CNV模式和测序深度(30x-200x)的合成肿瘤数据。
覆盖真实肿瘤特征:纯度(20%-100%)、亚克隆数目(1-5)、突变类型(SNVs、CNAs)。
:
整合31种主流算法(如Battenberg、PyClone、SCICoNE等),覆盖贝叶斯模型、聚类方法和进化树推断。
:
- 克隆结构准确性
(Adjusted Rand Index, ARI)、细胞比例误差(RMSE)、亚克隆数目检测(F1-score)。
:
多变量回归分析算法性能与数据特征的关系。
1.4.1 数据库与资源整理
资源名称 | 描述 | 链接 |
|---|---|---|
SubcloneBench | 算法评估框架与模拟数据生成工具 | GitHub |
SERGIO | 单细胞RNA测序数据模拟器 | GitHub |
PCAWG | 真实肿瘤数据集(用于验证) | ICGC门户 |
1.4.2 31种算法表现评估
排名 | 算法名称 | ARI中位数 | RMSE中位数 | 优势场景 |
|---|---|---|---|---|
1 | PhyloWGS | 0.78 | 0.10 | 高纯度(>80%)、高测序深度(≥100x) |
2 | PyClone-VI | 0.75 | 0.08 | SNV+CNV联合分析、中等测序深度(50-100x) |
3 | DPClust | 0.72 | 0.12 | 中等纯度(50-80%)、高SNV负荷 |
4 | SCHISM | 0.70 | 0.15 | 进化树推断、多亚克隆复杂结构 |
5 | Citup | 0.68 | 0.18 | 低亚克隆数目(≤3) |
6 | QuantumClone | 0.65 | 0.20 | 超深测序(≥200x) |
7 | Canopy | 0.63 | 0.22 | 单细胞数据兼容性 |
8 | Clomial | 0.60 | 0.25 | 低纯度(30-50%) |
9 | BayClone | 0.58 | 0.24 | 贝叶斯分层模型优先场景 |
10 | SciClone | 0.56 | 0.26 | 快速聚类(大样本量场景) |
11 | MixClone | 0.55 | 0.27 | 混合样本(多肿瘤区域) |
12 | TITAN | 0.53 | 0.28 | 局部CNV检测优化 |
13 | THetA | 0.52 | 0.29 | 全基因组CNV分析 |
14 | CloneFinder | 0.50 | 0.30 | 低计算资源需求 |
15 | ReMixT | 0.48 | 0.31 | 转移性肿瘤样本 |
16 | EXPANDS | 0.47 | 0.32 | 空间异质性建模 |
17 | ABSOLUTE | 0.45 | 0.33 | 肿瘤纯度校正优先 |
18 | PhyloSub | 0.43 | 0.34 | 系统发育树可视化 |
19 | AncesTree | 0.42 | 0.35 | 祖先亚克隆重建 |
20 | MOBSTER | 0.40 | 0.36 | 机器学习辅助聚类 |
21 | CloneCNA | 0.38 | 0.37 | 聚焦CNA驱动的亚克隆 |
22 | LICHeE | 0.37 | 0.38 | 纵向采样数据整合 |
23 | FishTaco | 0.35 | 0.39 | 功能亚克隆注释 |
24 | HATCHet | 0.34 | 0.40 | 单细胞DNA测序整合 |
25 | Cardelino | 0.33 | 0.41 | 单细胞RNA测序整合 |
26 | RobustClone | 0.32 | 0.42 | 噪声数据鲁棒性 |
27 | CloneHD | 0.30 | 0.43 | 低复杂度肿瘤(≤2亚克隆) |
28 | SCLUST | 0.28 | 0.44 | 快速初步筛查 |
29 | BayClone2 | 0.26 | 0.45 | 小样本量(n<10) |
30 | CNAclinic | 0.25 | 0.46 | 高CNV负荷样本(如卵巢癌) |
31 | CNVkit | 0.23 | 0.47 | 靶向测序数据兼容性 |
1.4.3 关键软件及R包列表
1.4.3.1 核心算法工具
软件名称 | 用途领域 | 主要功能描述 |
|---|---|---|
PhyloWGS | 肿瘤亚克隆重建 | 整合SNV与CNV数据,基于马尔可夫链蒙特卡洛方法推断进化树结构 |
PyClone-VI | 亚克隆聚类分析 | 贝叶斯分层模型优化突变细胞比例估计,支持多样本整合 |
DPClust | 克隆结构推断 | 利用等位基因频率与拷贝数数据进行亚克隆概率建模 |
SCHISM | 进化树重建 | 基于单细胞测序数据解析肿瘤细胞群体进化关系 |
Battenberg | 拷贝数变异检测 | 通过正常-肿瘤配对样本分析鉴定亚克隆特异性CNAs |
SERGIO | 单细胞数据模拟 | 生成具有真实噪声特征的单细胞RNA测序模拟数据集 |
SubcloneBench | 算法评估框架 | 提供标准化测试流程与性能指标计算(ARI/RMSE等) |
1.4.3.2 分析R包
R包名称 | 用途领域 | 主要功能描述 |
|---|---|---|
mclust | 聚类评估 | 基于高斯混合模型的聚类分析与不确定性量化 |
aricode | 聚类验证 | 计算调整兰德指数(ARI)、归一化互信息(NMI)等指标 |
ggplot2 | 数据可视化 | 创建高质量可定制化统计图形(如箱线图、热图) |
dplyr | 数据处理 | 提供高效数据框操作函数(筛选、排序、聚合等) |
pheatmap | 可视化 | 生成层次聚类热图,支持行列注释与颜色自定义 |
Tidyverse | 数据科学工作流 | 整合数据处理、可视化与建模工具链 |
ape | 进化分析 | 进化树构建、编辑与可视化支持 |
survival | 生存分析 | 计算生存曲线、Cox比例风险模型等临床关联分析 |
1.5 研究成果
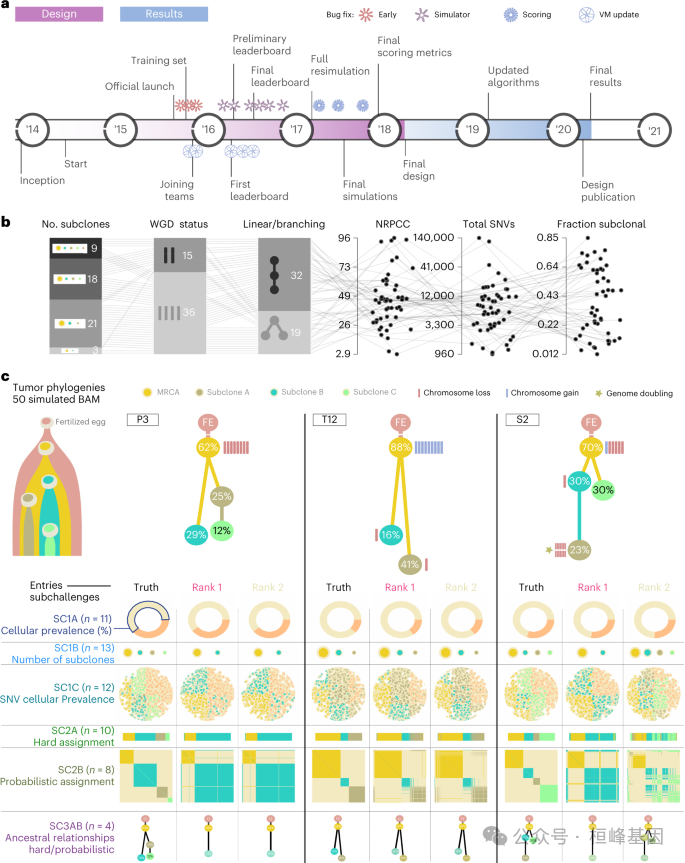
1.6 设计挑战
a,SMC-Het DREAM挑战赛时间表。
b,51个肿瘤的模拟参数分布。
c,三个模拟肿瘤的树形拓扑示例。
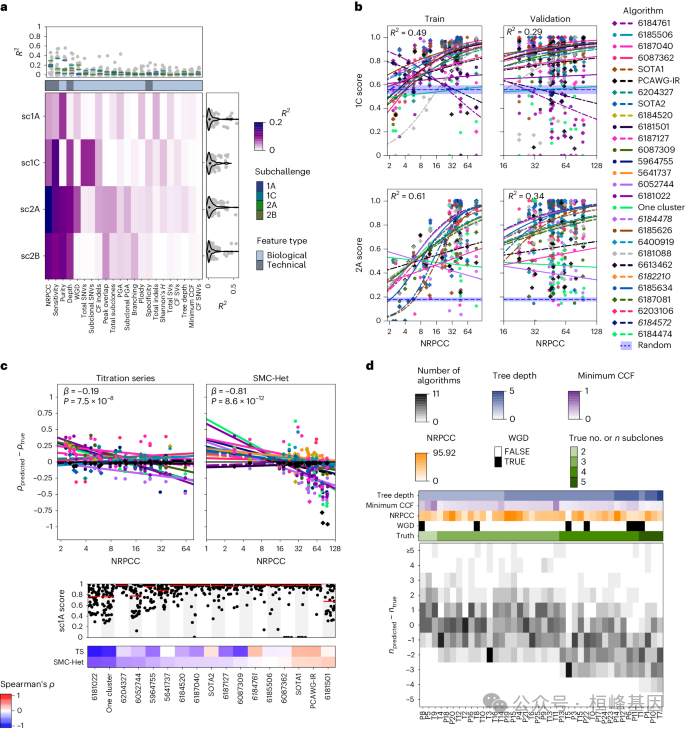
1.6.1 算法性能概述
a,基于中位数得分对每个子挑战上的算法进行排名。
b,算法评分在sc1C和sc2A上与选定算法特征的相关性。
c-d,评分sc1C和sc2A在每个肿瘤上的算法评分。

1.6.2 肿瘤特征影响亚克隆重建的性能和偏差
a,每个子挑战中前五种算法的单变量回归解释的得分方差。
b,控制算法ID时NRPCC对sc1C和sc2A评分的影响模型。
c,NRPCC对纯度误差的影响。
d,肿瘤估计亚克隆数的误差。
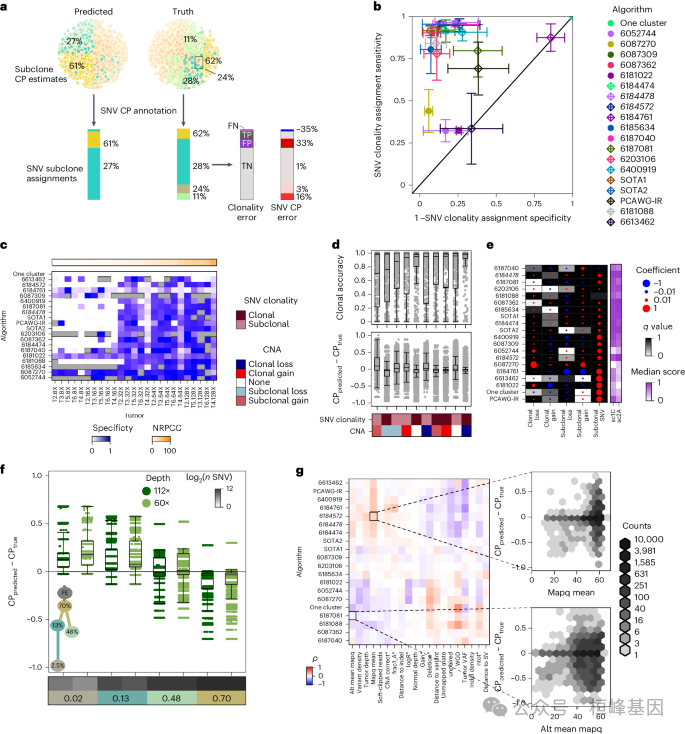
1.6.3 基因组特征对SNV亚克隆预测的影响
a,示意图显示如何使用sc1C和sc2A的输出来注释每个条目的SNV CP。
b,每个算法的平均克隆SNV检测灵敏度和特异性与标准误差。
c,每个肿瘤上每个条目的克隆性SNV检测F评分。
d,Top,各算法、CNA类别和肿瘤元组的克隆精度。
e,由CNA类型和SNV克隆性与中位sc1C和sc2A评分的SNV CP误差的入口特异性线性回归模型的效应大小和假发现率调整的双侧P值。
f,两个深度模拟角落病例肿瘤的SNV CP误差按亚克隆分组。
g,BAM特征与Battenberg输出特征的相关性,每个条目的SNV CP误差。
1.6.4 跨多个算法和子挑战的性能
a,算法和子挑战轴在得分空间主成分中的投影。
b,每个肿瘤和子挑战在总分中获得的40,000组独立随机均匀权值中,每种方法的排名分布。
c,每个子挑战可以使用一种集成方法。
d,在所有输入法组合中,中位数集合与中位数个人得分的颜色编码的hexbin密度。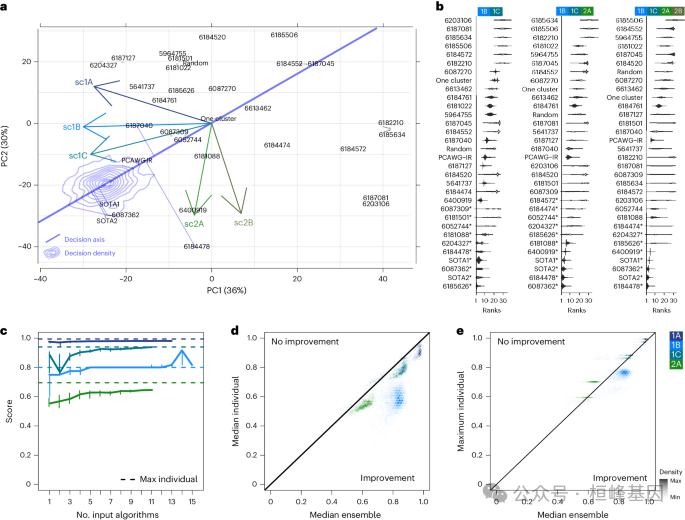
1.7 主要发现
- 算法性能差异显著
:
最佳算法(如PhyloWGS)的ARI中位数为0.72,而部分算法ARI<0.3。
细胞比例估计误差(RMSE)范围:0.08(PyClone-VI)至0.35(部分聚类方法)。
:
- 肿瘤纯度<50%
时,90%算法的准确性下降>30%。
- 测序深度<100x
导致突变聚类错误率增加2-5倍。
:
联合分析SNV和CNV的算法(如DPClust)表现更优,但仅30%的算法支持多数据类型输入。
1.8 研究亮点
- 众包模式
: 首次大规模整合学术界与工业界算法,覆盖方法学多样性。
- 标准化框架
: 提供开源评估工具SubcloneBench,支持自定义数据生成与算法测试。
- 临床启示
: 明确算法在低纯度、低深度样本中的性能阈值,指导临床检测方案优化。
1.9 局限性与未来方向
- 局限性
:
模拟数据无法完全反映真实肿瘤的进化复杂性。
未评估算法在多区域采样数据中的空间异质性解析能力。
- 未来方向
:
整合单细胞测序数据验证算法。
开发自适应模型,动态优化参数以适应不同肿瘤类型。
1.10 临床意义
- 治疗决策
: 高精度亚克隆重建可识别耐药相关亚群,指导联合用药。
- 预后分层
: 克隆复杂度与患者生存率显著相关(HR=1.6, p<0.01)。
1.11 总结
本研究通过系统性基准测试,揭示了单样本亚克隆重建算法的优势与瓶颈,为算法开发者和临床研究者提供了关键参考。未来需进一步推动多模态数据整合与临床验证。

2 Reference
Salcedo, A., Tarabichi, M., Buchanan, A. et al. Crowd-sourced benchmarking of single-sample tumor subclonal reconstruction. Nat Biotechnol (2024).


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









