







简 介
单细胞RNA测序(scRNA-seq)是一种发现和注释细胞类型和状态的通用工具,但细胞亚型的确定和注释往往是主观的和任意的。通常甚至不清楚给定的群集是否均匀。在这里提出了一个基于熵的统计,ROGUE以准确地量化鉴定细胞团的纯度并证明ROGUE指标是广泛适用的,并且能够在广泛的模拟和真实数据集上对聚类纯度进行准确,敏感和稳健的评估。将这一指标应用于成纤维细胞、B细胞和大脑数据,确定了额外的亚型,并演示了ROGUE指导分析在特定亚群中检测精确信号的应用。ROGUE可以应用于所有已测试的scRNA-seq数据集,对于评估假定簇的质量、发现纯细胞亚型以及构建全面、详细和标准化的单细胞图谱具有重要意义。

软件包安装
软件包安装非常简单,利用devtools安装即可。
install.packages("tidyverse")
if (!requireNamespace("devtools", quietly = TRUE))
install.packages("devtools")
devtools::install_github("PaulingLiu/ROGUE")数据读取
数据输入需要两种文件:一种是count矩阵,一种是meta信息。
suppressMessages(library(ROGUE))
suppressMessages(library(ggplot2))
suppressMessages(library(tidyverse))
expr <- readr::read_rds(file = "./DC.rds.gz")
meta <- readr::read_rds(file = "./info.rds.gz")
expr[1:5, 1:4]
## _p1t1__bcGDSJ _p1t1__bcDRQX _p1t1__bcFPXB _p1t1__bcHVVV
## A2M 0 0 0 0
## A2ML1 0 0 0 0
## AAAS 0 0 0 0
## AACS 0 0 0 0
## AAGAB 0 0 0 0
dim(expr)
## [1] 9795 913
head(meta)
## # A tibble: 6 × 26
## Patient Tissue `Barcoding emulsion` Library Barcode `Total counts`
## <chr> <chr> <chr> <chr> <chr> <dbl>
## 1 p1 tumor p1t p1t1 bcGDSJ 4731
## 2 p1 tumor p1t p1t1 bcDRQX 1212
## 3 p1 tumor p1t p1t1 bcFPXB 2639
## 4 p1 tumor p1t p1t1 bcHVVV 2978
## 5 p1 tumor p1t p1t1 bcGJVN 1509
## 6 p1 tumor p1t p1t1 bcFSSY 3369
## # ℹ 20 more variables: `Percent counts from mitochondrial genes` <dbl>,
## # `Most likely LM22 cell type` <chr>, `Major cell type` <chr>, ct <chr>,
## # used_in_NSCLC_all_cells <lgl>, x_NSCLC_all_cells <lgl>,
## # y_NSCLC_all_cells <lgl>, used_in_NSCLC_and_blood_immune <lgl>,
## # x_NSCLC_and_blood_immune <dbl>, y_NSCLC_and_blood_immune <dbl>,
## # used_in_NSCLC_immune <lgl>, x_NSCLC_immune <lgl>, y_NSCLC_immune <lgl>,
## # used_in_NSCLC_non_immune <lgl>, x_NSCLC_non_immune <lgl>, …实例操作
通常,在无监督的scRNA-seq数据分析中,甚至不清楚给定的簇是否均匀。cluster 纯度的概念,并引入了一个概念新颖的统计,称为ROGUE,来检查一个给定的集群是否是一个纯细胞群体。
过滤掉低丰度基因和低质量细胞
matr.filter()过滤掉低丰度的基因和低质量的细胞。
expr <- matr.filter(expr, min.cells = 10, min.genes = 10)表达式熵模型
为了应用S-E模型,我们使用SE_fun函数计算每个基因的表达熵。
ent.res <- SE_fun(expr)
head(ent.res)
## # A tibble: 6 × 7
## Gene mean.expr entropy fit ds p.value p.adj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 LYZ 1.65 0.762 1.27 0.510 0 0
## 2 HLA-DQB2 1.35 0.569 1.01 0.437 0 0
## 3 BIRC3 1.21 0.458 0.886 0.428 0 0
## 4 HSPA1A 1.54 0.766 1.17 0.406 0 0
## 5 HLA-DRB1 2.99 2.24 2.59 0.353 0 0
## 6 GZMB 1.26 0.586 0.931 0.345 0 0S-E模型能准确识别信息基因
使用SEplot函数来可视化S和E之间的关系。
SEplot(ent.res)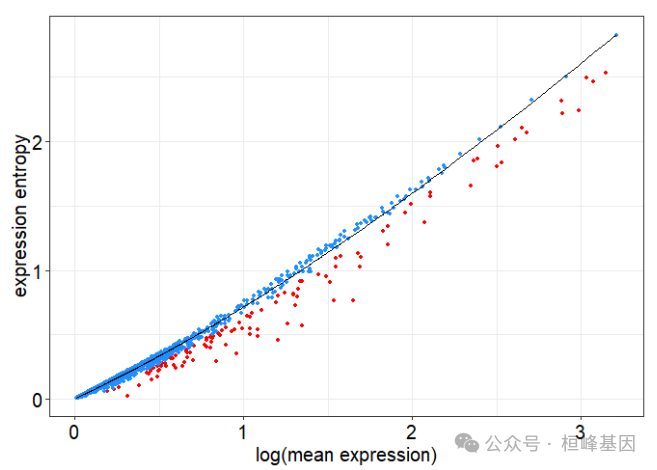
ROGUE计算
为了评估这个DC种群的纯度,使用CalculateRogue函数计算ROGUE值。该群体的ROGUE值为0.72,从而证实了其异质性。
rogue.value <- CalculateRogue(ent.res, platform = "UMI")
rogue.value
## [1] 0.7219202计算每个样本的每个假定聚类的ROGUE值
为了准确估计每个簇的纯度,建议计算不同样本中每种细胞类型的ROGUE值。
rogue.res <- rogue(expr, labels = meta$ct, samples = meta$Patient, platform = "UMI",
span = 0.6)
rogue.res
## tDC2 tpDC tDC3 tDC1
## p1 0.8376831 0.8604547 0.8494896 0.8481964
## p2 NA NA NA NA
## p3 0.8028900 0.8941508 0.8995863 0.9150546
## p4 0.8041421 0.8992421 0.8763108 0.8658948
## p5 0.8702724 0.9321946 0.9247687 NA
## p6 0.8596472 NA 0.8892388 0.9280764
## p7 0.9262411 0.9028763 0.8949111 0.9419589ROGUE 可视化
箱线图
rogue.boxplot(rogue.res)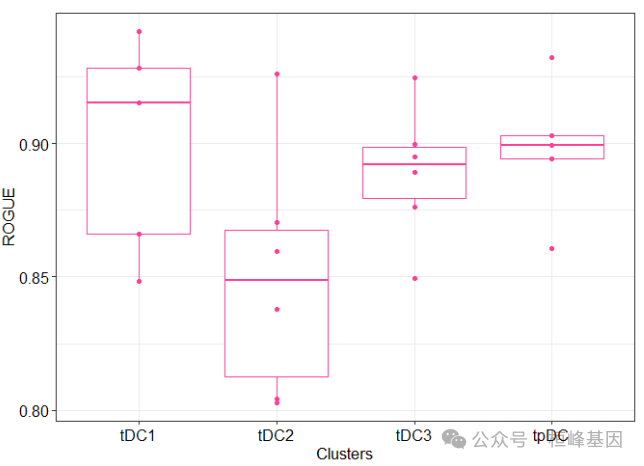
rogue.res
## tDC2 tpDC tDC3 tDC1
## p1 0.8376831 0.8604547 0.8494896 0.8481964
## p2 NA NA NA NA
## p3 0.8028900 0.8941508 0.8995863 0.9150546
## p4 0.8041421 0.8992421 0.8763108 0.8658948
## p5 0.8702724 0.9321946 0.9247687 NA
## p6 0.8596472 NA 0.8892388 0.9280764
## p7 0.9262411 0.9028763 0.8949111 0.9419589热图
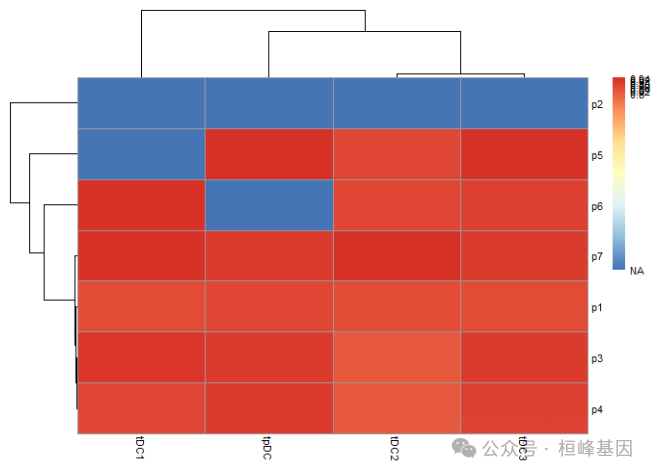
绘制热图的时候我们发现结果里面带有NA这种情况,有两种解决办法,一种就是将NA赋值较大的值,一种就是将数据做成对称的形式,这里我们就用简单粗暴的方式。
rogue.res[is.na(rogue.res)]=-0.8
pheatmap::pheatmap(rogue.res,
legend_breaks = c(-0.8,seq(from=0.8, to=1, by=0.02)),
legend_labels = c("NA",seq(from=0.8, to=1, by=0.02)),
legend = TRUE,
fontsize = 8
# filename = 'pheatmaps.pdf'
)
Reference
Liu B, Li C, Li Z, Wang D, Ren X, Zhang Z. An entropy-based metric for assessing the purity of single cell populations. Nat Commun. 2020 Jun 22;11(1):3155.
细胞生信分析教程
桓峰基因公众号推出单细胞生信分析教程并配有视频在线教程,目前整理出来的相关教程目录如下:
SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)
SCS【6】单细胞转录组之细胞类型自动注释 (SingleR)
SCS【7】单细胞转录组之轨迹分析 (Monocle 3) 聚类、分类和计数细胞
SCS【8】单细胞转录组之筛选标记基因 (Monocle 3)
SCS【9】单细胞转录组之构建细胞轨迹 (Monocle 3)
SCS【10】单细胞转录组之差异表达分析 (Monocle 3)
SCS【11】单细胞ATAC-seq 可视化分析 (Cicero)
SCS【12】单细胞转录组之评估不同单细胞亚群的分化潜能 (Cytotrace)
SCS【13】单细胞转录组之识别细胞对“基因集”的响应 (AUCell)
SCS【15】细胞交互:受体-配体及其相互作用的细胞通讯数据库 (CellPhoneDB)
SCS【16】从肿瘤单细胞RNA-Seq数据中推断拷贝数变化 (inferCNV)
SCS【17】从单细胞转录组推断肿瘤的CNV和亚克隆 (copyKAT)
SCS【18】细胞交互:受体-配体及其相互作用的细胞通讯数据库 (iTALK)
SCS【21】单细胞空间转录组可视化 (Seurat V5)
SCS【22】单细胞转录组之 RNA 速度估计 (Velocyto.R)
SCS【24】单细胞数据量化代谢的计算方法 (scMetabolism)
SCS【25】单细胞细胞间通信第一部分细胞通讯可视化(CellChat)
SCS【26】单细胞细胞间通信第二部分通信网络的系统分析 (CellChat)
SCS【28】单细胞转录组加权基因共表达网络分析(hdWGCNA)
SCS【29】单细胞基因富集分析 (singleseqgset)
SCS【30】单细胞空间转录组学数据库(STOmics DB)
SCS【31】减少障碍,加速单细胞研究数据库(Single Cell PORTAL)
SCS【32】基于scRNA-seq数据中推断单细胞的eQTLs (eQTLsingle)
SCS【33】单细胞转录之全自动超快速的细胞类型鉴定 (ScType)
SCS【34】单细胞/T细胞/抗体免疫库数据分析(immunarch)
SCS【35】单细胞转录组之去除双细胞 (DoubletFinder)
SCS【36】单细胞转录组之k-近邻图差异丰度测试(miloR)
SCS【39】单细胞转录组之降维散点图的美化 (SCpubr)
SCS【40】单细胞转录组之便捷式细胞类型注释 (scMayoMap)
SCS【41】基于贝叶斯反卷积法整合分析bulk和scRNA-seq (BayesPrism)
SCS 42.基于单细胞转录组表型数据构建临床预测模型 (Sicssor)
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
桓峰基因官网正式上线,请大家多多关注,还有很多不足之处,大家多多指正!
http://www.kyohogene.com/
桓峰基因和投必得合作,文章润色优惠85折,需要文章润色的老师可以直接到网站输入领取桓峰基因专属优惠券码:KYOHOGENE,然后上传,付款时选择桓峰基因优惠券即可享受85折优惠哦!https://www.topeditsci.com/

















 444
444

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









