







问题描述:使用Qiime2生成的feature table(特征表),注释文件以及代表性序列中的ASV名称往往是很长的一段数字(如:“84b577b185f76483981b055fc7c878cb”),在后续的分析中可能会与某些软件不兼容。我们可以用如下代码将这些ASV的特征名称修改为ASV1、ASV2、ASV3……这样的形式。
修改特征表(feature table)和分类表(taxonomy table)处理基本的表格即可,这里的关键问题是:如何读取序列文件,并修改序列名称,R包seqinr可以实现这一问题。具体R代码如下:
#change OTU ID in the feature table
feature.table2 <- read.delim("yourfeature-table.txt")
oldID <- feature.table2$OTU.ID
newID <- paste("ASV",1:length(feature.table2$OTU.ID),sep = "")
feature.table2$OTU.ID <- newID
View(feature.table2)
write.table(feature.table2,
"your-feature-table2.txt",
quote = FALSE,
row.names = FALSE,
col.names = TRUE,
sep = "\t")
#Change feature ID in taxonomy table
tax <- read.delim("your-taxonomy.tsv")
names(newID) = oldID
newFeatureID <- newID[tax$Feature.ID]
tax$Feature.ID <- newFeatureID
View(tax)
write.table(tax,
"your-taxonomy2.tsv",
quote = FALSE,
row.names = FALSE,
col.names = TRUE,
sep = "\t")
#Change the sequence ID in fasta file
library(seqinr)
seq <- read.fasta(file = "your-dna-sequences.fasta",
as.string = TRUE,
seqtype = "DNA",
forceDNAtolower = FALSE)
oldSeqID <- names(seq)
newSeqID <- newID[oldSeqID]
#names(seq) <- newSeqID
#seq[1:5]
write.fasta(sequences = seq,names = newSeqID,file.out="your-dna-sequences2.fasta")
效果如下:
feature-table修改前:
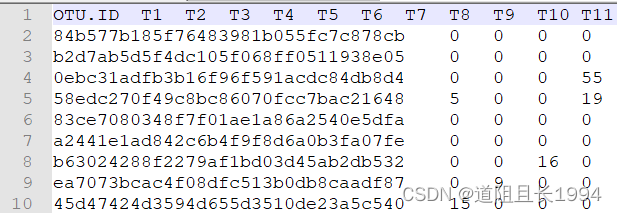
feature-table修改后:
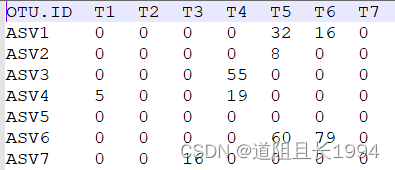
注释文件修改前:
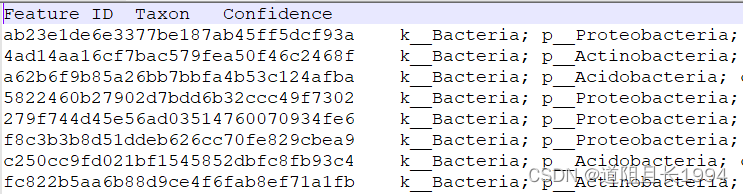
注释文件修改后:
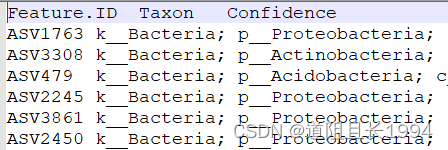
代表性序列修改前:
代表性序列修改后:

值得一提的是,我们还可以用“seqinr”包对序列的格式(每行的字符数)进行修改,以适应某些特殊软件的需要,效果如下:
代表性序列格式修改前:

代表性序列格式修改后:

代码如下:
library(seqinr)
seq <- read.fasta(file = "your-dna-sequences.fasta",
as.string = FALSE,
seqtype = "DNA",
forceDNAtolower = FALSE)
oldSeqID <- names(seq)
newSeqID <- newID[oldSeqID]
#names(seq) <- newSeqID
#seq[1:5]
write.fasta(sequences = seq,
names = names(seq),
file.out="C:/Users/hutianlong/Desktop/your-dna-sequences2.fasta",
nbchar = 50)
序列格式修改因人而异,文章中难以尽述,有相关需求的朋友可以CSDN私信我哈。


















 5605
5605

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











