









 RoseTTAFold是一个基于深度学习的蛋白质结构预测方法,利用多序列比对(MSA)和自注意力机制。它通过1D序列、2D距离图和3D坐标信息的转换和整合,构建了一个三轨神经网络。论文中介绍了从MSA中提取协同进化信息、使用轴向注意力处理MSA和更新配对特征的步骤,以及如何通过SE(3)-Transformer细化3D结构。此外,它还展示了与传统方法的比较,证明了其在准确性上的优势。
RoseTTAFold是一个基于深度学习的蛋白质结构预测方法,利用多序列比对(MSA)和自注意力机制。它通过1D序列、2D距离图和3D坐标信息的转换和整合,构建了一个三轨神经网络。论文中介绍了从MSA中提取协同进化信息、使用轴向注意力处理MSA和更新配对特征的步骤,以及如何通过SE(3)-Transformer细化3D结构。此外,它还展示了与传统方法的比较,证明了其在准确性上的优势。
(1)论文地址:点击下载
(2)官方代码:https://github.com/RosettaCommons/RoseTTAFold.
(3)训练数据集:
采用HHblits对UniRef30和BFD序列数据库进行迭代搜索同源序列。序列搜索的E-value截止值逐渐放宽,直到生成的MSA至少有2000个覆盖率为75%的序列,或者5000个覆盖率为50%的序列(均为90%序列标识截止值)。
然后根据生成的MSA通过HHsearch方法在PDB100数据库中搜索模板信息。
HHblits和HHsearch是HH-suite中的两个方法,比同类型的方法速度要快很多。
(4)如何下载sequence和structure databases(UniRef30,BFD,PDB100)
UniRef30:http://wwwuser.gwdg.de/~compbiol/uniclust/2020_06/UniRef30_2020_06_hhsuite.t ar.gz
BFD:https://bfd.mmseqs.com/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt.tar.gz
PDB: https://files.ipd.uw.edu/pub/RoseTTAFold/pdb100_2021Mar03.tar.gz
目录
8、基于pair features中编码的结构信息更新MSA特征:
1、摘要
该方法在AlphaFold的基础上,通过对1D sequence、2D distance map和3D coordinate进行依次转换和整合而提出了一个“three-track neural network”。该网络还能够通过序列信息快速生成准确的蛋白质-蛋白质结构,缩短了传统方法的时间。
2、highlights
-
对于1D sequence MSAs派生的逆协方差矩阵;
-
用注意力机制代替2D卷积,能够更好地表示序列上距离较远的残差之间的相互作用;
-
使用two-track网络体系结构,其中1D sequence和2D distance map的信息进行迭代转换并来回传递;
-
使用SE(3)-等变Transformer网络直接精炼由双轨网络生成的原子坐标;
-
采用一种端到端学习,从最终生成的三维坐标,通过反向传播,通过所有网络层返回到输入序列,从而优化所有网络参数。
总体的流程图如下图所示:
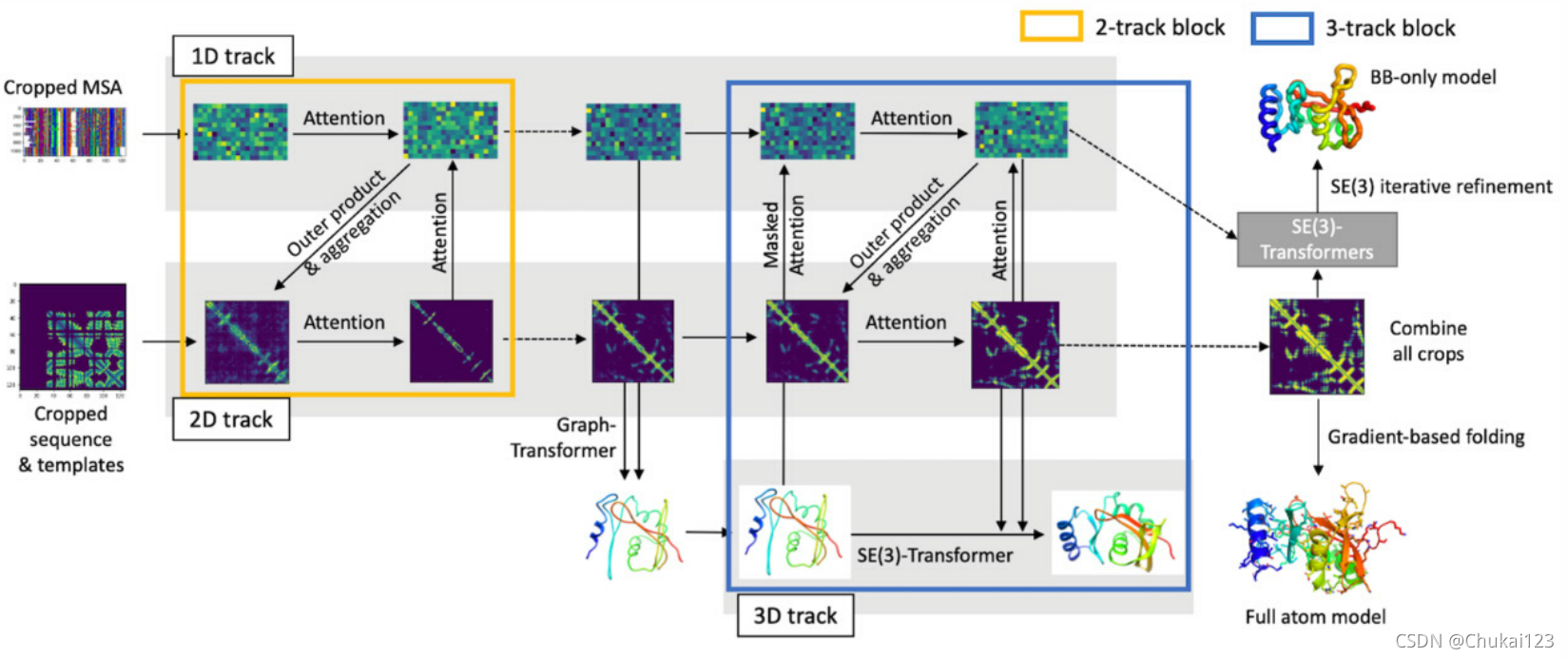
3、1D track:
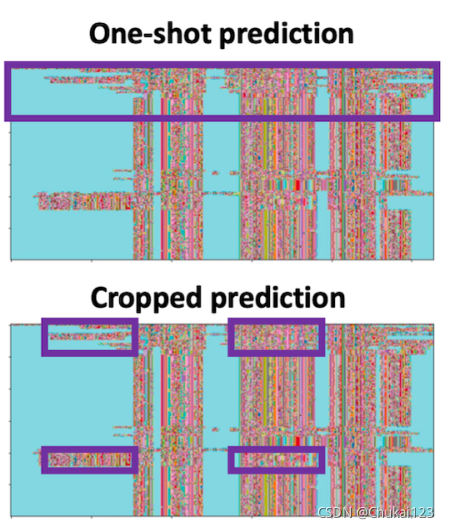
首先是cropped MSA(裁剪的MSA),这个操作主要测试了多个不连续的序列(cropped MSA)比一次性预测(uncropped MSA)整个结构更加准确;但是这个裁剪也不是随便裁剪的,主要就是删除一些最不相关的序列;同时也会加快模型运行的速度;(减少内存限制)
4、Initial Embedding:
将cropped MSA作为输入,它的输入格式为:[N×L],其中N表示MSA中有多少条序列,L表示序列的长度(序列对应氨基酸的每个位置)。将初始的MSA进行标记化(tokenized),用于进一步处理。得到氨基酸和间隙被称为字符级标记(总共21个)。然后得到一个维度为Dmsa,经过这个序列化操作以后经过一个Embedding层;
在上述的Embedding中又采用了sinusoial positional encoding(正弦位置编码,使用正余弦函数表示绝对位置(不同位置之间存在一定的约束关系),通过两者乘积得到相对位置),重点关注每个序列的残基(residues)(就是对应上述MSA中输入格式中的L),可以使得网络在学习过程中重点关注。然后对于序列维度,增加了一个查询序列的指示器,因为MSA是主要关注查询序列意外的信息。
多序列比对(多序列联配,Multiple sequence alignment, MSA)
是指把多条(3 条或以上)有系统进化关系的蛋白质分子的氨基酸序列
或核酸序列进行比对,尽可能地把相同的碱基或氨基酸残基排在同一列上。
这样做的意义是,对齐的碱基或氨基酸残基在进化上是同源的,
即来自共同祖先(common ancestor)。Templates:
从模板中获取对齐位置的两两距离和方向,以及1D的信息(位置相似度和对齐置信评分)和标量特征(HHsearch概率、序列相似度和序列标识)。这两个特性都是通过沿着2D输入的两个轴平铺而连接到2D输入的。模板首先通过一轮轴向注意(行式注意接着列式注意),然后使用像素式注意机制合并成单一的2D特征矩阵。处理后的特征矩阵然后与二维平铺查询序列连接,然后映射到隐藏维(dpair)以获得成对特征。同时对2D特征矩阵也增加了正弦位置编码。
5、通过自注意力机制处理MSA:
(1)自注意力机制是注意力机制的变体,其减少了对外部信息的依赖,更擅长捕捉数据或特征的内部相关性。
(2)自注意力机制在文本中的应用,主要是通过计算单词间的互相影响,来解决长距离依赖问题。经过上述的Embedding之后,得到的MSA特征的形状:[N×L×d],其中N表示序列的数量,L表示序列的长度,d表示嵌入的维度。
然后对该2D特征进行轴向注意方法处理(就是分别对行和列交替注意)
单独使用Row Attention(或者Col Attention),即使是堆叠好几次,也是无法融合全局信息的。
一般来说,Row Attention 和 Col Attention要组合起来使用才能更好的融合全局信息。然后采用Performer架构作为列注意(就是序列的维度 ),从而减少内存(O(LN^2)-->O(LN))。
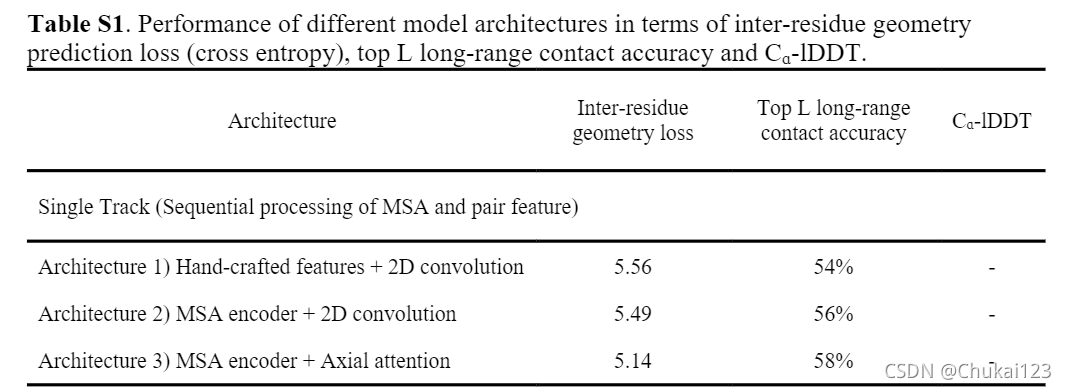
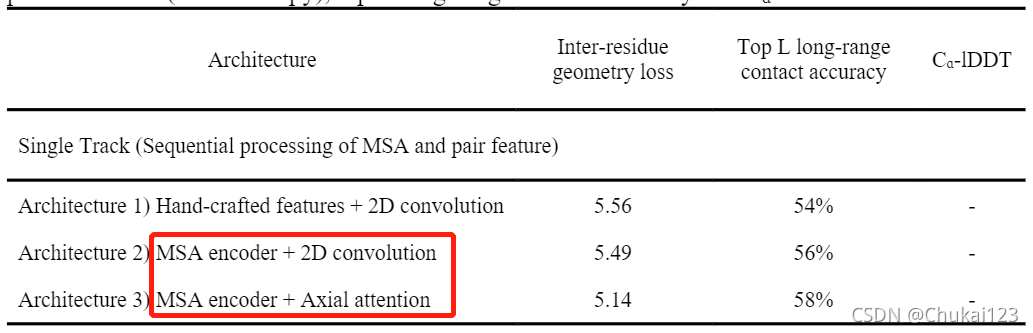
Preformer的时间、空间复杂度。实验表明,注意力加速和内存减少几乎是最优的。然后文中对比了MSA编码器与协同进化提取器(Architecture 1)与具有手工制作特征(Architecture 2)(序列轮廓和逆协方差矩阵)进行了比较,如下表所示:Architecture 1优于Architecture 2
对行注意力(对于残基的维度,L维度),文中对比了两种方法:(1)un-tied attention(2)softly tied-attention(MSA transformer)。
其中softly tied-attention提出的主要原因在于,针对同源序列应该具有相似的结构。然后引入了学习的位置权重因子组合MSA序列中的注意信号从而修改这种tied-attention,以减少未对齐区域的贡献。
tied-attention的公式如下所示:
![]()
对应的算法流程:
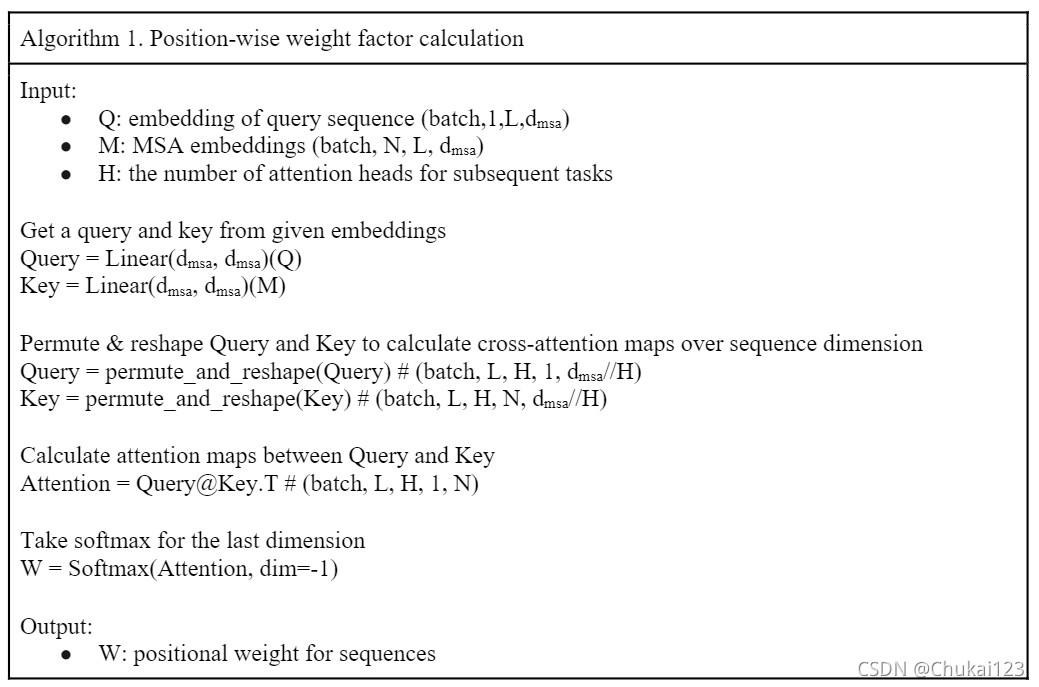
其中N表示MSA中的序列数量,Qn和Kn是第N个输入序列的查询和键矩阵,Wn是对应序列的位置权重因子。
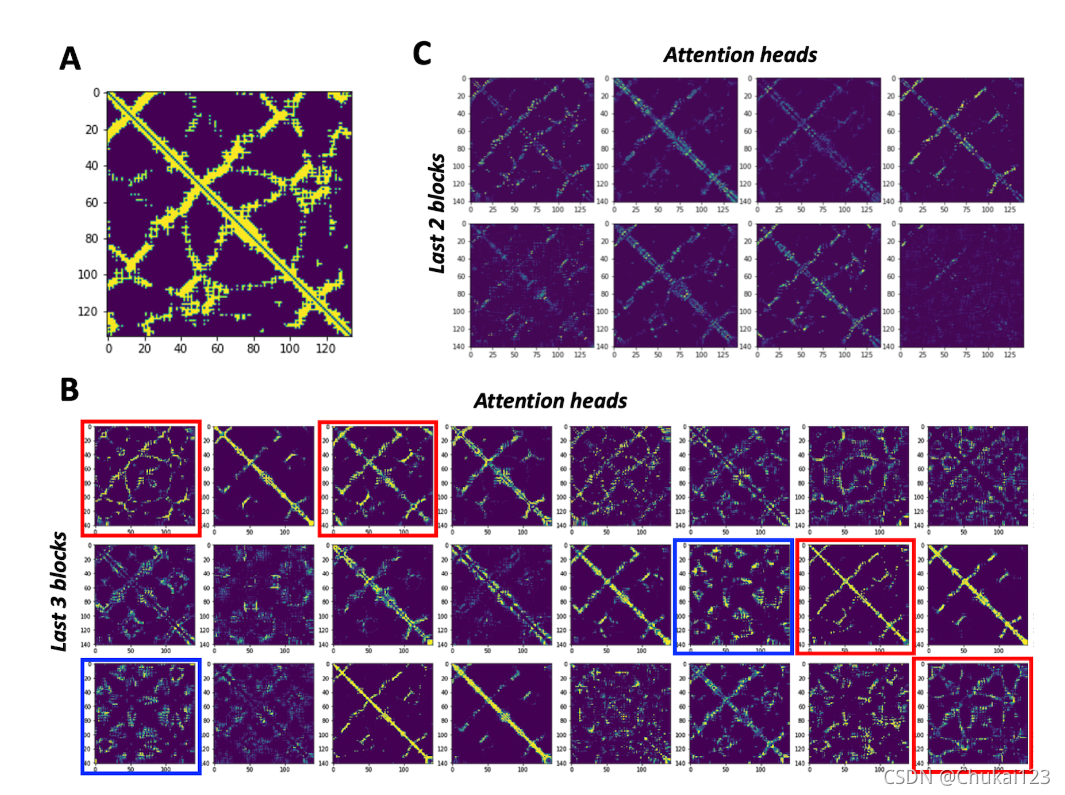
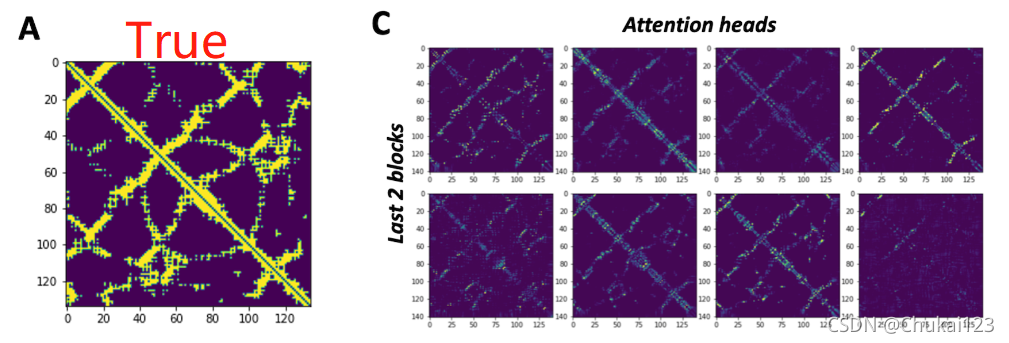
采用tied-attention不仅提高了预测的性能,而且在残基注意图谱中显示了与真实接触图谱的相关性
![]()
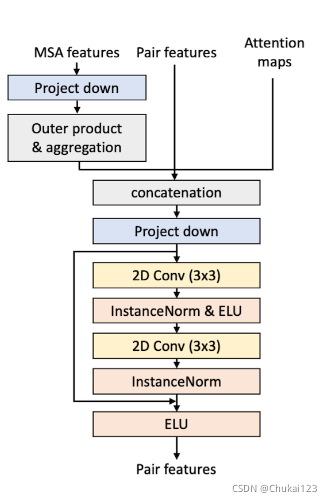
6、使用来自MSA特征的协同进化信息更新配对特征:
为了从给定的MSA特征中提取成对残基对相互作用信息,文中采用了CopulaNet方法中的外积和聚合的思想。
其中外积:可以提取每个序列中两个残基之间的相关性;而聚合:则是使用平均池化聚合从所有同源蛋白质获得的外积,最后产生两个残基之间共进化的度量。
文中并没有详细说明,查看了一下CopulaNet的原文:

以一个one-hot编码方式为例:
每个序列的位置对就会生成一个21×21的替换表(原始的氨基酸个数是20,这里增加了一个间隙,因此是21),然后从所有序列中取替换表的平均值时,得到的21×21特征会显示不同的分布,这就取决于它们是否在三维空间中相互作用。
如果在21×21的特征分布比较广泛,即使在三维空间中也无法表明两个残基有空间接触;如下图:

如果聚合的特征存在比较突出的分布(表示相关突变),在三维空间中就存在更高的交互机会;如下图(随便找的图)

上述的one-hot编码只是一个例子说明,文中并没有采用one-hot,而是采用MSA embedding的方式;
采用outer product会增加内存的负载,因此文中将MSA embedding映射到一个32的隐藏维度。(降维?)然后从MSA中的每个序列中提取任意两个残基的embedding,再使用位置序列权重计算所有序列的外积加权平均值。
然后将这些聚合的共同进化的特征与1D特征(MSA特征的加权平均)和来自先前MSA更新的残基注意图谱相结合。然后将结合后的特征映射到成对特征的隐藏尺寸(这个是之前的32?)。
为了更好的结合新提取的成对特征和之前的成对特征,文中测试了两种方法:(1)直接在前馈网络后面添加两个成对特征;(Architecture 5)(2)拼接两对特征,然后再经过一个2D卷积网络的残差块。(Architecture 6),方法(2)的方法效果明显更好

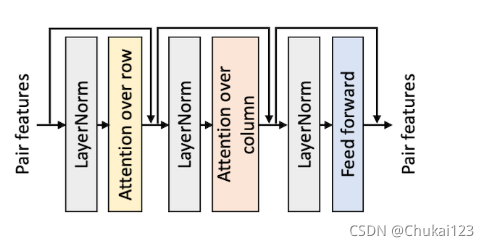
7、通过行和列方向的自注意力细化成对特征:
对于上一步中来自MSA的共同进化信号的更新的pair features,采用轴向注意力(如下图,首先是row,然后是column)替代卷积,实验验证轴向注意力明显优于卷积(如下图)
然后考虑到内存的使用,于是采用Performer架构(scale-up),很大程度上减少了内存的使用,实验验证如下所示:
![]()
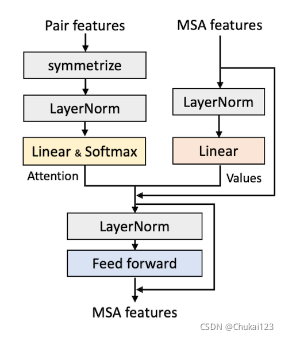
8、基于pair features中编码的结构信息更新MSA特征:
因为在AlphaFold2中最显著的特征就是通过pair features更新MSA特征,本文中提出了两种方法
通过pair features更新MSA特征:
(1)将pair features到MSA的更新看作一种encoder-decoder的过程,在MSA和pair features之间采用cross-attention。
(2)将pair feaures中的attention应用于MSA特征(成为direct-attention)。(3D空间中靠近的位置来更新MSA特征)。
文中也进行对应的验证:direct-attention优于cross-attention
![]()
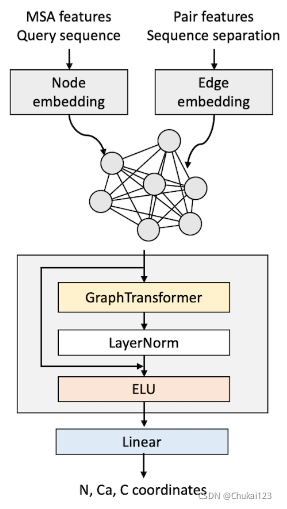
9、初始3D结构预测:
采用Graph Transformer的架构作为初始的主干网络,输入为一个完全连接的图,而其中的节点表示蛋白质中的残基。
节点和边缘embeddings分别从平均 MSA 特征与one-hot编码查询序列和pair features以及序列分离相结合中学习。主干网络采用使用四个Graph Transformer的堆叠进行评估,然后通过简单的线性变换来预测每个残基节点的N,Ca,C原子的笛卡尔坐标。
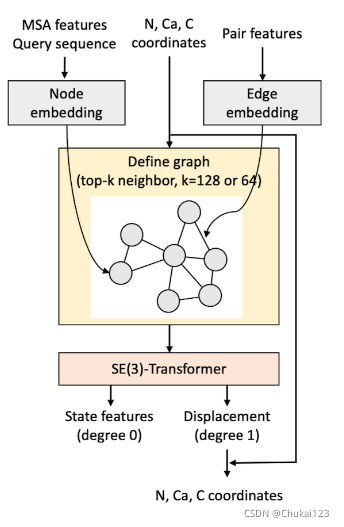
10、通过SE(3)-Transformer更新结构:
SE(3)-Transformer 用于最新的 MSA 和 3-Track模型中的pair features来细化给定的 3D (N、Ca、C)坐标。蛋白质图由Ca原子的节点定义,每个节点连接到k个近邻。N原子和C原子的位置通过将位移向量包含到对应对应的Ca原子作为一阶节点特征来编码(向量节点特征)。
来自更新的MSA和one-hot编码的Query sequence的节点特征嵌入(Node embedding)作为标量节点特征。
还有pair features也包含在SE(3)-Transformer的输入特征中;
SE(3)-Transformer预测了Ca原子的位移以及N和C原子到更新的Ca位置的新位移矢量。同时提供了标量节点特征(此处称为状态特征,用于下一节中描述的基于结构的MSA的更新)。
11、基于3D结构更新MSA特征:
和基于2D-track模型中pair features的MSA更新类似,MSA特征基于从当前3D结构导出的注意图谱进行更新。
根据上面的状态特征计算出四个注意图谱,并根据具有四个不同截止点(8,12,16和21Å )和Ca距离对它们进行Mask(即总流程图中的Masked Attention),以便它只关注3D空间中的邻居。
相同的注意图谱同样用于MSA中的所有序列,逐点前馈层(pointwise feed-forward)进一步处理来自Masked多头注意力的输出。过程如下图所示:
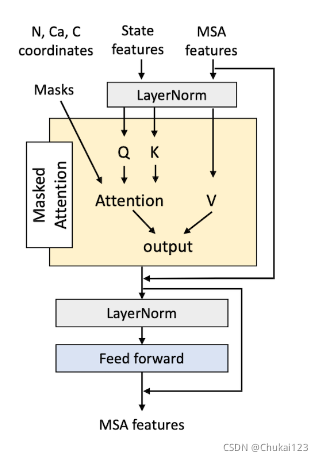
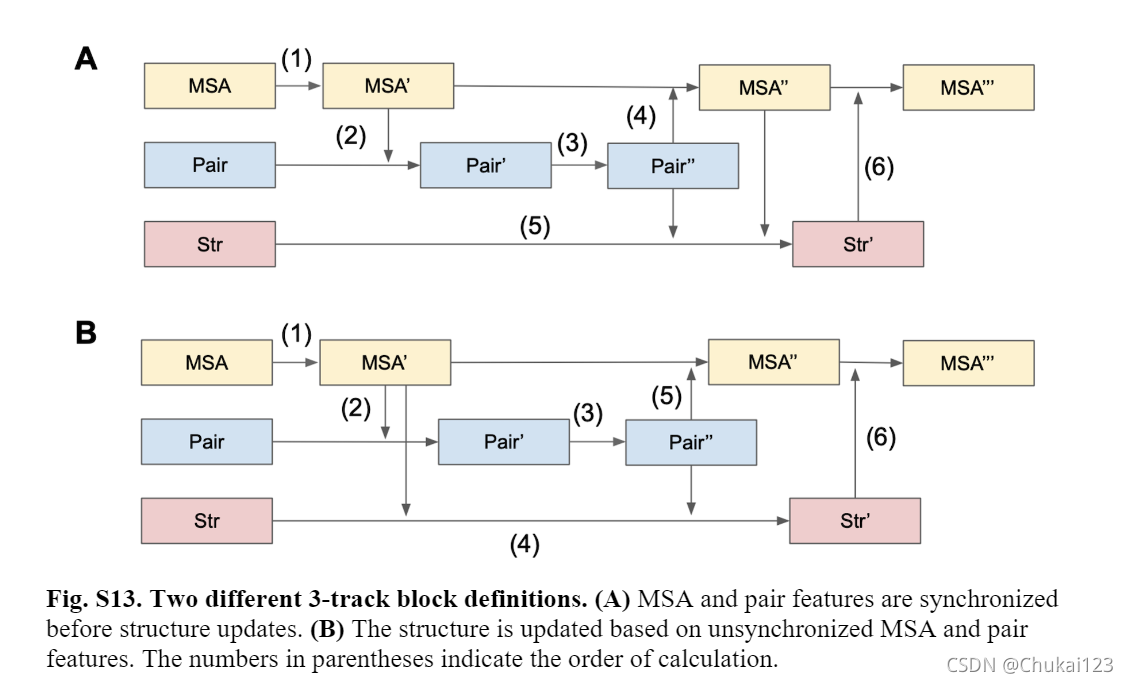
12、2D-track和3D-track特征处理块的定义:
总的流程图中的黄色框中四个箭头表示2D-track的流程;
首先通过自注意力更新MSA特征,从MSA中提取共同进化特征,并将其和之前的pair features相结合;然后通过轴向注意力进一步优化pair features特征,并基于当前pair features中编码的结构信息更新MSA特征;
总的流程图中的蓝色框中箭头过程表示3D-track的流程:
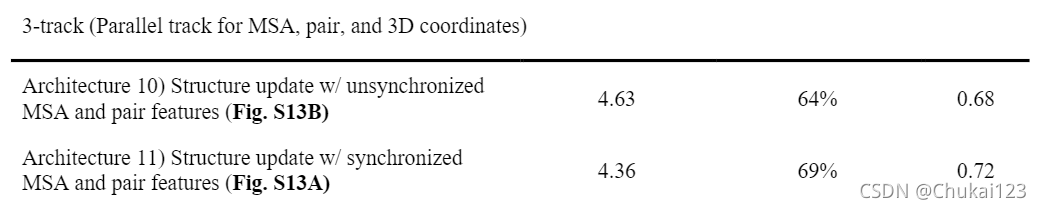
3D-track中流通顺序很重要,文中验证了两种不同的方式来交流1D、2D和3D-track。在同步MSA和pair features之前和之后更新结构。如下图B所示:
基于同步MSA和pair feature的3D坐标更新的性能表现最优,如下表所示:
13、残基对距离和方向预测:
通过一个单独的残差块(由两个3×3大小的2D卷积),然后跟一个1D卷积(1×1)和softmax激活预测inter-residue几何表示。因为Cᵦ-Cᵦ 距离的图谱和沿假设序列的二面角Cᵦ-Cᵦ 键是对称的,我们通过使用转置和未转置特征映射的平均值作为这些预测的输入,在网络中强制对称。
14、通过网络进行迭代优化的附加结构模块:
虽然结构在3D-track中明确采样,但引入了一个额外的结构模块,以基于组合的1D特征和2D残差几何预测构建模型,用于多重cropps的推断。
主干网络中N、Ca、C原子的初始坐标是使用简单的基于图形的架构(见上文的初始3D结构预测部分)生成的,节点和边缘特征来自平均MSA特征以及2D距离和方向分布。
然后用于生成初始坐标的相同节点和边缘特征,使用多个SE(3)-Transformer层进一步细化这些坐标。在SE(3)-Transformer层的末端,根据来自最终SE(3)-Transformer层的状态特征来评估残差的Ca-1DDT。
在训练过程中,文中没有使用任何迭代,通过网络的单次传递优化了参数。在推理时将最终SE(3)-Transformer层的输出坐标作为输入输入到第一个SE(3)-Transformer层,使用该结构模块作为迭代细化工具,如下图所示:预测出的Ca-1DDT作为评分函数,以决定何时停止迭代并从所有结构中选择最终模型。
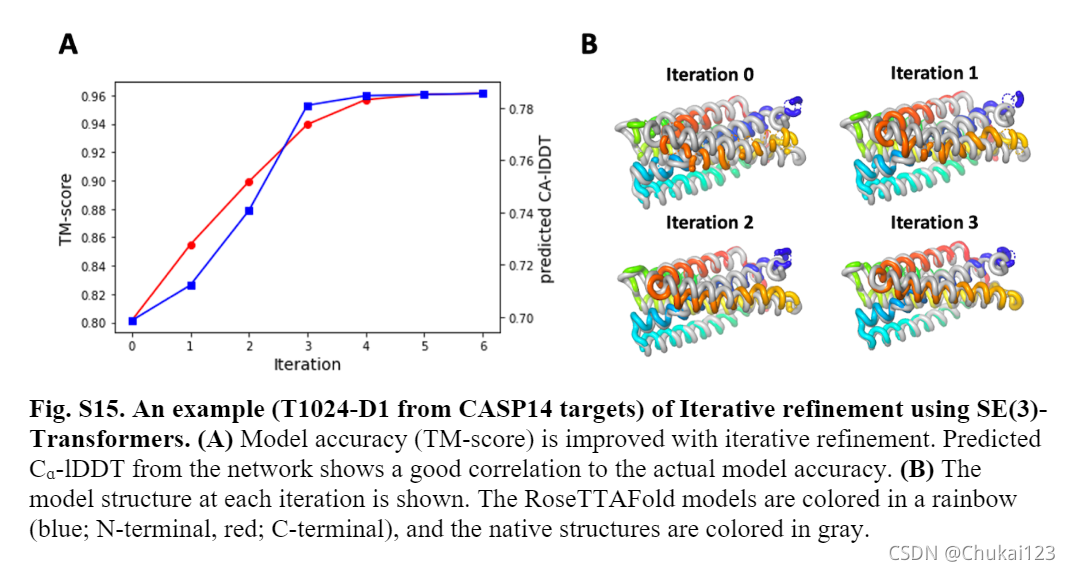
15、2-track端到端模型与3-track模型的比较:
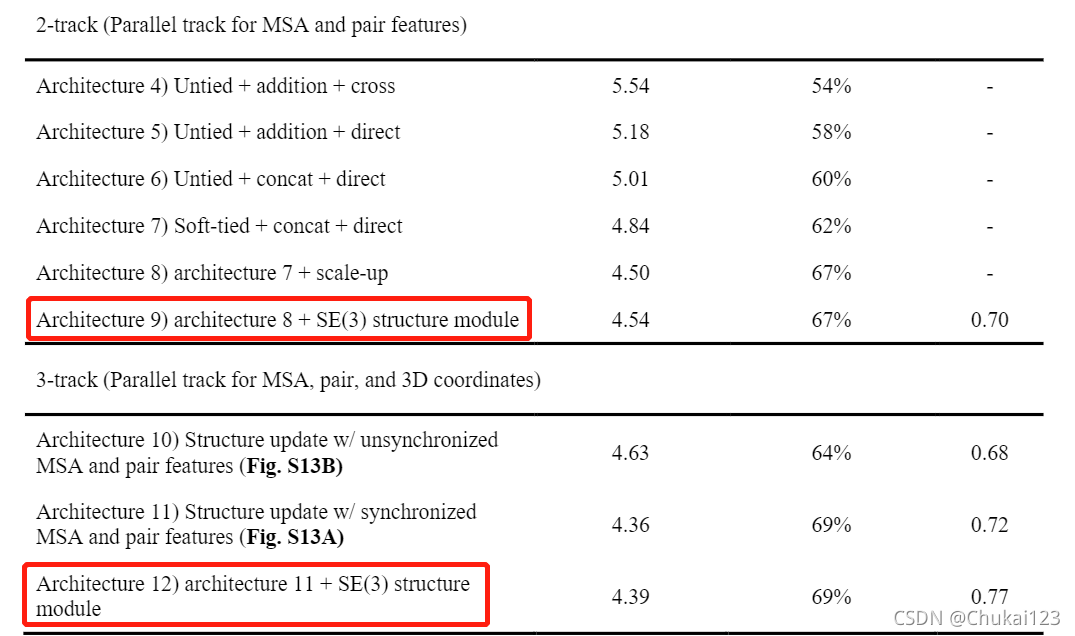
通过实验验证2-track端到端模型不如3-track端到端模型如下表和下图所示:
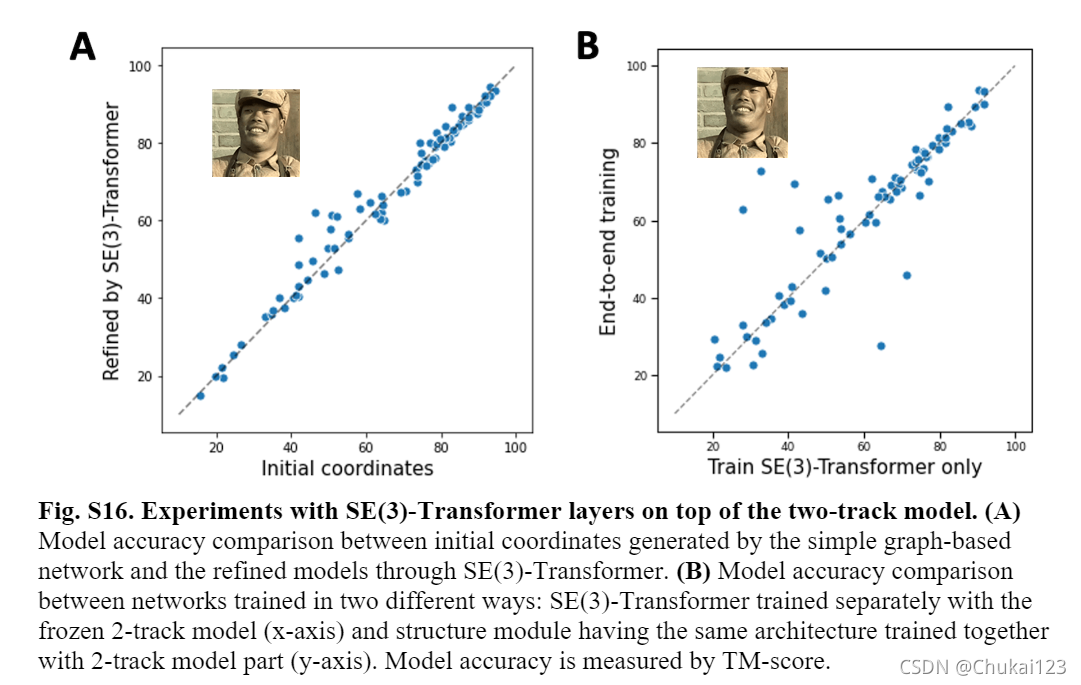
ps:上述只是针对Rosettafold的整个流程进行了探索,具体的实验部分,可以查阅原文!


















 456
456

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











