






目录
二、将linux产生文件导入windows,设定Rstudio工作目录
一、DESeq2包安装
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("DESeq2")二、将linux产生文件导入windows,设定Rstudio工作目录
通过共享文件夹/mnt/hgvfs复制SRR3418005_count.txt和SRR3418006_count.txt
在windows上新建G:/R/TEST1作为工作目录
setwd("G:\\R\\TEST1")
#确认目前工作目录
getwd()

安装包勾选DESeq2
 三、从个别计数文件开始(整合、过滤、DESeq分析)
三、从个别计数文件开始(整合、过滤、DESeq分析)
从HTSeq输出的计数文件,如SA01.txt,SA02.txt,SA03.txt,SA04.txt
创建一个CSV文件sampleinfo.csv,并放在目录下
通过DESeq2包在R上整合,如下

3.1整合表达矩阵,定义列名为文件名
# 定义文件编号
file_numbers <- c(01, 02,03,04)
# 格式化文件编号为2位数字符串
formatted_numbers <- sprintf("%02d", file_numbers)
# 生成完整的样本编号
samples <- paste0("SA", formatted_numbers)
# 定义一个函数来读取和处理每个样本文件
read_sample_file <- function(sample_id) {
fname <- paste0(sample_id, ".txt")
data <- read.delim(fname, header=FALSE, row.names=1)
return(data)
}
# 使用lapply读取所有样本文件,结果存储在一个列表中
list_of_samples <- lapply(samples, read_sample_file)
# 检查所有样本文件是否具有相同的行数
num_rows <- sapply(list_of_samples, nrow)
if (!all(num_rows == num_rows[1])) {
stop("Not all files have the same number of rows.")
}
# 使用do.call和cbind将列表中的所有数据框按列合并,coun_table可以替换自己想输出的名字
count_table <- do.call("cbind", list_of_samples)
# 设置合并后的数据框的列名为样本编号
colnames(count_table) <- samples
3.2读取分组信息,定义factor和对照组
#从sampleinfo.csv读取grpdata的信息,确保sampleinfo提前建立好,sample和dex信息一一对应
grpdata <- read.csv("sampleinfo.csv", header = T)
#定义组基于grp dex这一列信息,将 dex 列转换为因子类型,同时设定水平顺序(Etoh_8h为第一水平),这样Etoh_8h为对照组
grpdata$dex <- factor(grpdata$dex,levels = c("Etoh_8h", "ABA_8h")) 对照组一定要设置正确,levels后第一个“”组内是对照组,后面计算就是基于factor的levels
3.3过滤,表达矩阵改列名为条件信息
这一步也可以不更换列名,结果图就以SA01的方式展示结果,或者输入
colnames(d1) <- grpdata$sample
#从count_table中选择那些至少有3个值大于3的行,并将这些行保存到新的数据框d2中,通过DESeq整合的方式第一列(gene_id)不参与计算
d1 <- count_table[rowSums(count_table > 3) > 2,]
#设置d1的列名,行名由grpdata$dex决定
colnames(d1) <- grpdata$dex3.4DEseq计算(关键步骤)
#创建DESeqDataSet对象,design =~dex表示你希望基于dex这一列信息
d1.deseq <- DESeqDataSetFromMatrix(countData = d1, colData = grpdata, design =~dex)
#使用DESeq函数对d.deseq对象进行差异表达分析
d1.deseq <- DESeq(d1.deseq)
#使用results函数提取treated列中"ABA_8h"和"Etoh_8h"(对照组、写在后面)这两个水平,比较它们在差异表达上的变化
results_ABA_8h_vs_Etoh_8h <- results(d1.deseq, contrast=c("dex", "ABA_8h", "Etoh_8h"))
# 找出 padj 小于 0.05 的基因索引
padj_indices <- which(results_ABA_8h_vs_Etoh_8h$padj < 0.05)
# 找出 log2FoldChange 的绝对值大于等于 1.5 的基因索引
log2FoldChange_indices <- which(abs(results_ABA_8h_vs_Etoh_8h$log2FoldChange) >= 1.5)
#找出同时满足上述两个条件的基因索引
sig_indices <- intersect(padj_indices, log2FoldChange_indices)
#使用这些索引来提取满足所有条件的显著基因
sig1 <- results_ABA_8h_vs_Etoh_8h[sig_indices, ]
#创建表格,包含sig1的所有信息(padj,log2FoldChange)
write.csv(sig1,file = "sig1.csv")打开csv,

四、从现有的整合计数表开始
确保merged.count.txt在文件夹中且整合了数据

merged.counts <- read.delim("merged.count.txt", sep="\t")- 使用
read.delim函数读取一个名为"merged_count.txt"的文本文件。 sep="\t"参数指定了文件中的数据是以制表符(tab)分隔的。这是read.delim函数的默认行为,因此实际上这个参数是可选的,除非你的文件不是用制表符分隔的。- 将读取的数据框(data frame)赋值给变量
merged.counts。
五、可视化检查潜在的离群值、样本差错
5.1 热图
#使用getVarianceStabilizedData函数获取方差稳定化后的数据,这通常用于可视化或进一步的分析
vsd1<- getVarianceStabilizedData(d1.deseq)
#创建了一个热图来展示方差稳定化后数据的相关性
heatmap(cor(vsd1),cexCol=0.75,cexRow=0.75)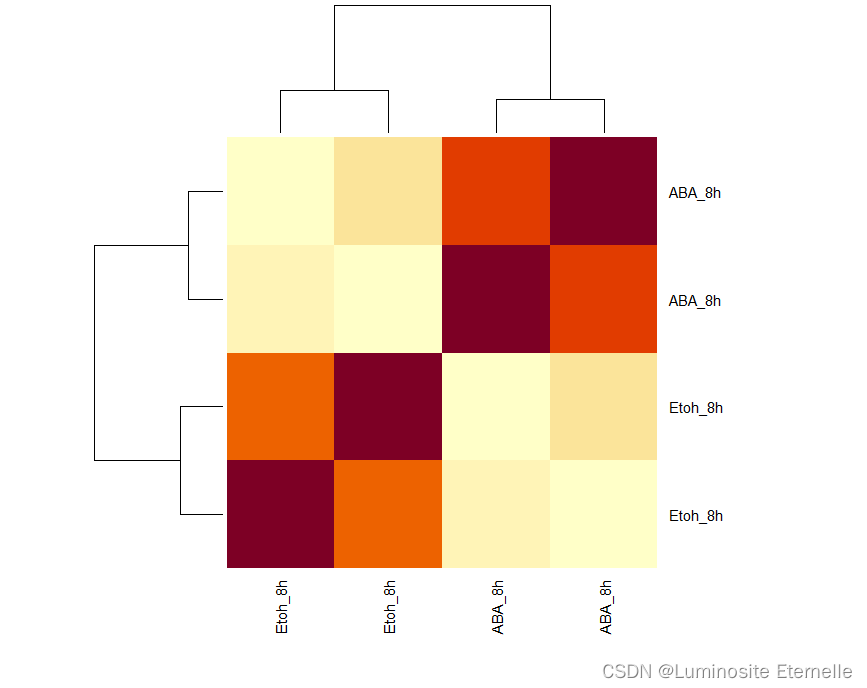

5.2 PCA图
#以PCA图展示
#prcomp是R中用于主成分分析的一个函数。t(vsd)是对vsd数据框进行转置操作
pr <- prcomp(t(vsd))
#绘制主成分分析的结果图
#pr$x包含了主成分得分,即每个观测值(在这个情况下是样本)在主成分空间中的坐标。
#col="white"设置点的颜色为红色,这通常用于确保标签清晰可见(因为文本标签通常是黑色的)。
#main="PC plot"设置图的主标题为“PC plot”。
#xlim=c(-22,15)设置了x轴的范围
plot(pr$x,col="red",main="PC plot",xlim=c(-100,100))
#添加文本标签
#pr$x[,1]和pr$x[,2]分别提取第一主成分和第二主成分的得分。
#labels=colnames(vsd)设置标签为vsd数据框的列名,因为在对转置后的数据进行PCA后,我们实际上想要显示的是原始vsd数据框的列名(即样本名)
#cex=0.7设置文本大小缩放因子为0.7,用于调整标签大小。
text(pr$x[,1],pr$x[,2],labels=colnames(vsd),cex=0.7)

PCA可以看到根据主成分PC1样本被分成2个不同的组
5.3 plotPCA图
# 对数据进行 VST 转换
vst1 <- vst(d1.deseq, nsub=500)
# 绘制 PCA 图
plotPCA(vst1, intgroup = "dex")















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









