







低共熔溶剂(DESs)是由一定的氢键供体(HBDs)和氢键受体(HBAs)按照一定比例混和组成的低共熔混合物,常见的DESs是由氯化胆碱与不同配体按照一定摩尔比形成的,主要由于氯化胆碱价格便宜、来源广泛、无毒且可生物降解。低共熔溶剂的物理化学性质对于DESs的应用至关重要,大量研究结果证实不同的氢键供受体具有不同的物化性质。因此低共熔溶剂作为一种新型绿色溶剂,在矿物分选、生物质催化转化、无机功能材料制备以及电化学等方面已经得到了广泛的应用。而对于低共熔溶剂的分子模拟主要在于氢键供体与氢键受体相互作用。因此,运用Materials Studio软件对于胆碱类低共熔溶剂中的氯化胆碱(ChCl)与丙二酸(MA)进行模型搭建与计算。
主要步骤如下:1.利用Materials Studio画图面板画出氯化胆碱与丙二酸分子模型(如图1),并应用Dmol3进行模型优化得到最稳定的优化构型并计算前线分子轨道性质(如图2)以及态密度(如图3)。 2.将氯化胆碱与丙二酸放入同一面板中应用Dmol3进行相互作用计算得到氯化胆碱-丙二酸低共熔溶剂分子构型(ChCl-MA)(如图4),得到稳定构型后可以进行径向分布函数性质分析(如图5)。
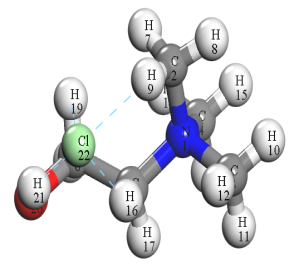
ChCl
MA
图1氯化胆碱、丙二酸优化构型
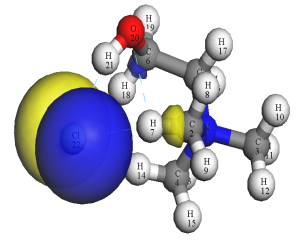
ChCl-HOMO轨道 ChCl-LUMO轨道 MA HOMO轨道
MA LUMO轨道
图2 前线分子轨道
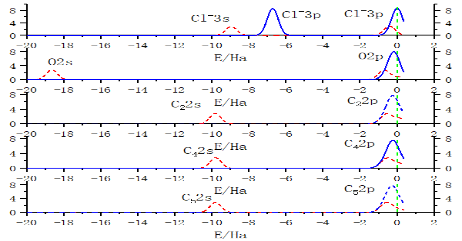
图3 ChCl分波态密度
图4 ChCl-MA稳定构型
由图1可以看出氯化胆碱中的氯离子和胆碱阳离子之间以氢键相互作用。由图2可以看出氯化胆碱中失电子活性位点位于氯离子上,而得电子活性位点聚集胆碱基团上。其中胆碱阳离子上得电子位点主要分布在C-H上。丙二酸主要失电子位点为一个C=O上的O,得电子主要分布在C、O上。通过图3可以看出氯离子与氧、碳原子发生强烈共振,最高占据态内是由Cl-3s、Cl-3p、O-2s、O-2p、C2-2s、C2-2p、C4-2s、C4-2p、C5-2s、C5-2p贡献,其中Cl-3p、O-2p、C2-2p、C4-2p、C5-2p占主导地位,说明形成的p-p杂化轨道是阴阳离子间电子转移的主要通道。图4为经过优化得到的氯化胆碱-丙二酸稳定构型,发现两种物质以
多重分子间氢键相结合,图5径向分布函数证明氢键供受体之间主要以分子间氢键相互作用。
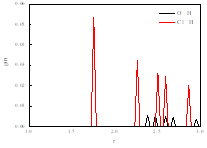
ChCl-MA径向分布函数
Dmol3计算面板:
最后,欢迎通过公众号"320科技工作室"联系我们


















 4535
4535

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











