










A chromosome-level genome assembly provides insights into Cornus wilsoniana evolution, oil biosynthesis and floral bud development
染色体级基因组组装为光皮梾木的进化、油脂生物合成和花芽发育提供了见解,光皮树基因组,多组学分析基本全覆盖~

摘要
光皮梾木 W. 是一种木本油料植物,含油量高且具有强大的降血脂效果,使其成为中国药用、园林景观和生态目的的宝贵物种。为了推进这一物种的遗传研究,我们利用PacBio和Hi-C数据创建了光皮梾木的草图基因组组装。基于一个锚定了11条染色体的染色体级组装,估计基因组大小为843.51 Mb。N50 contig大小和N50 scaffold大小分别计算为4.49 Mb和78.00 Mb。此外,注释了30,474个编码蛋白基因。比较基因组学分析显示,光皮梾木在大约12.46百万年前(Mya)与其最近的物种分化。此外,山茱萸科和Nyssaceae科之间的分化发生在>62.22百万年前。我们还发现了全基因组复制事件和全基因组三倍体γ事件,分别发生在大约44.90和115.86百万年前。我们进一步推断了染色体的起源,这揭示了光皮梾木染色体组型的复杂演化历史。通过转录和代谢分析,我们识别了两个可能在控制油酸与亚油酸比率中起关键作用的FAD2同源基因。我们进一步研究了代谢物与基因之间的关联,并确定了可能影响光皮梾木花形态的33个MADS-TF同源基因。总体而言,这项研究为未来旨在识别光皮梾木关键性状的遗传基础的研究奠定了基础。
介绍
光皮梾木基因组调查、组装和评估
光皮梾木 Wangerin(2n = 2x = 22,即Swida wilsoniana的同义词)是属于山茱萸属(Cornus)[1]的落叶灌木,是中国北温带54种多年生木本植物中的一种(Dogwoods names-Encyclopedia of Life)。山茱萸属的名称来源于拉丁语单词cornu,意为“角”,这是对植物坚硬木质的引用。种名wilsoniana是为了纪念植物学家欧内斯特·威尔逊(Ernest Wilson),他在20世纪初收集了这个特定物种。它通常被称为威尔逊的山茱萸。春天,这个物种会生产成簇的白色花朵,称为伞房花序。这些花随后会发展成早秋出现的紫黑色浆果。虽然其花朵和茎的颜色可能不如其一些更知名的亲戚那样引人注目,但它的剥落、斑驳的树皮在冬季尤其吸引人,因此它获得了“幽灵山茱萸”的俗名。在中国,它还被称为广皮树。鉴于其全年的观赏价值,光皮梾木是景观设计中的一个极佳选择,作为标本植物。
在中国,光皮梾木是一种具有重要生态和经济价值的树种。该植物的果实是球形的,直径在6.0至9.3毫米之间,含油量在10%至17%之间。此外,果实的果肉含有55-59%的油脂[2]。从光皮梾木果实中提取的食用油已经食用了一个多世纪。这种油主要由三种类型的脂肪酸组成,即棕榈酸(约15%)、油酸(约30%)和亚油酸(约40%)[3, 4]。最近的研究发现光皮梾木具有强大的降血脂效果,这归因于其高浓度的多不饱和脂肪酸,特别是亚油酸和γ-亚麻酸[3]。因此,光皮梾木果油可以作为一种均衡的饮食油,不仅因其降血脂功能,也因为它能克服必需脂肪酸的缺乏。
从植物中获取的脂肪和油类是人类饮食中的重要组成部分。过去几年中,人们对探索高油含量植物中油脂生物合成的基因组研究兴趣日益增加[5-12]。与常见的植物油相比,光皮梾木种子油的不饱和脂肪酸含量高于棕榈油,与花生、大豆和橄榄油相似,但低于茶油和菜籽油[13]。廖等人的研究表明,光皮梾木油的皂化值高于茶油和花生油,但低于大豆油。其过氧化值高于大多数其他含油植物,但低于花生。酸值高于大多数常见植物油,但低于茶油、大豆油和菜籽油[13]。总体而言,光皮梾木果油的理化性质表现出色。然而,目前缺乏光皮梾木染色体级参考基因组资源。因此,这个物种内油脂生物合成的基因组基础仍未得到充分理解。罗等人已经进行了关于果实发育过程中关键的脂肪酸去饱和酶7(FAD7)基因的初步调查[14]。他们揭示了基因表达的动态模式,但尚未解开分子调控机制。然而,在其他含油植物中,许多转录因子已被实验证明参与油脂生物合成[15-18]。例如,WRINKLED1 (WRI1)可以直接调控多个参与油脂生物合成的基因[19],B3结构域转录因子FUSCA3 (FUS3)触发各种脂肪酸生物合成基因的表达,如脂肪酸去饱和酶3 (FAD3)和脂肪酸延长酶1 (FAE1)[20]。为了填补wilsoniana的知识空白,需要进一步进行基因组研究,包括使用先进的基因组技术开发核染色体级参考基因组。这将使研究人员能够识别参与油脂生物合成的基因,并更好地理解这种重要植物物种中脂肪酸和油脂产生的分子机制。
基因组测序为研究物种演化、识别遗传变异和选择作物改良的理想特性提供了宝贵资源[21]。为了深入了解光皮梾木,我们利用PacBio和Hi-C数据生成了一个草图基因组。然后,我们对基因组特征进行了系统注释,如重复序列、非编码RNA (ncRNA)基因和编码蛋白基因。比较基因组学分析显示光皮梾木与山茱萸(Cornus controversa)有着密切相关的物种。它们都是山茱萸科的成员,并在12.46百万年前(Mya)分化。此外,山茱萸科和Nyssaceae科的分化发生在>62.22百万年前。我们还发现了全基因组复制(WGD)事件和全基因组三倍体γ(WGT-γ)的证据,分别发生在约44.90和115.86百万年前。此外,我们进行了染色体组成的演化分析,阐明了光皮梾木的染色体组成及其与原始染色体的对应关系。最后,我们调查了可能涉及油脂生物合成和花芽发育的潜在基因。特别是,我们发现脂肪酸去饱和酶2(FAD2)同源基因可能在控制油酸与亚油酸(O/L)比率中发振关键作用。我们进一步研究了代谢物与基因之间的相关性,并显示了33个MADS-TF同源基因可能影响光皮梾木的花形态。这些信息为这一重要植物种的性状改良和育种提供了宝贵的见解。总的来说,光皮梾木基因组的染色体级组装为未来对山茱萸科的演化研究和这一物种的基因组研究奠定了基础。基因组数据还将有助于识别负责关键农艺性状的基因,加速光皮梾木作为一种有价值的作物植物的育种和改良。
结果
光皮梾木基因组调查、组装和评估
为了构建光皮梾木的基因组,从中国广东采集了新鲜嫩叶(图1A)。光皮梾木的体细胞为二倍体,含有11对染色体,包括20条长的中部着丝粒染色体和2条短的近中部着丝粒染色体(图1B及补充数据表S1)。基于123倍覆盖度的Illumina短读序列和k-mer分析,光皮梾木的基因组大小估计为941.39 Mb(图1C及补充数据表S2)。进一步利用PacBio的单分子实时(SMRT)测序技术改善了基因组组装。最终的基因组组装包含529个contigs,contig N50为4.49 Mb,最大长度为23.07 Mb,总长904.62 Mb(表1)。这个组装大小与基于k-mer分析估计的941.39 Mb基因组大小一致(图1C及补充数据表S3)。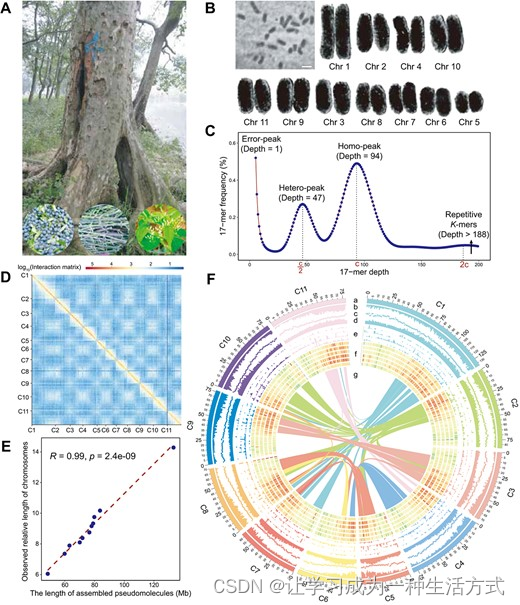
光皮梾木的高质量组装和基因组特征 A. 光皮梾木的照片。底部的三张照片从左到右分别显示了果实、藤蔓和嫩叶。 B. 光皮梾木的核型。标尺为1微米。单独的染色体被数字提取用于比较。 C. 光皮梾木基因组大小估计和多态性的k-mer分布。'c'是同源峰的k-mer深度。 D. 基于Hi-C测序捕获的染色体构象的11个拟染色体的组装。分辨率为500 kb。 E. 组装长度与所有染色体观察到的物理长度之间的相关性。 F. 使用1-Mb宽的区块跨越11条染色体描绘基因组特征。
-
轨道a, 11条染色体。
-
轨道b, 基因密度(每1 Mb范围1-92)。
-
轨道c, GC含量(每1 Mb范围31-39%);方向朝外。
-
轨道d, 转座元素(TE)密度(每1 Mb范围293-1281)。
-
轨道e, ncRNA密度(每1 Mb范围0-10);从外到内依次是miRNA、rRNA、snRNA和tRNA。
-
轨道f, 读取密度(每1 Mb范围0-5.8);数据通过log10转换并取生物重复的平均值;从外到内依次是叶子、高产芽、低产芽、高产果实、低产果实和花。
-
中心g, 曲线连接自WGD事件以来可能保留的同源区域。
Table 1
Statistics for the 光皮梾木 genome assembly.
| Contig level (Illumina + PacBio) | Scaffold level (Illumina + PacBio + Hi-C) | Chromosome level (Illumina + PacBio + Hi-C) | |
|---|---|---|---|
| Total number of contigs/scaffolds | 529 | 356 | 11 |
| Total length of sequences (bp) | 904 624 738 | 904 654 138 | 843 505 137 |
| Shortest sequence length (bp) | 6642 | 6642 | 48 466 712 |
| Longest sequence length (bp) | 23 068 660 | 133 174 022 | 133 174 022 |
| Contig/scaffold N50 size (bp) | 4 490 562 | 78 005 221 | 78 005 221 |
| Contig/scaffold N90 size (bp) | 965 924 | 48 466 712 | 59 859 763 |
| Total number of Ns | 20 | 29 414 | 29 414 |
| GC (%) | 35.63 | 35.63 | 35.58 |
| Number of genes | 31 640 | 31 610 | 30 474 |
我们采用Hi-C技术将contigs组装成拟染色体(补充数据表S2)。共843.51 Mb(93.24%)的序列成功锚定到了11个拟染色体上(图1D和补充数据图S1)。有345个contigs,总共61.12 Mb,未能锚定到11个拟染色体上。然而,我们的分析显示,这些未锚定的345个contigs应被视为冗余的基因组序列,而非来自外部来源的污染物(补充数据图S2)。最终,scaffold N50为78.00 Mb,最大染色体长度为133.17 Mb,从而得到总共904.65 Mb的高质量scaffold级基因组组装(表1)。我们通过比较体细胞中11组染色体的物理长度与组装长度来验证组装的准确性,并发现它们是一致的(图1E)。这11条染色体的大小范围从59.86 Mb到133.17 Mb不等(图1F),每个染色体仅含有1500到5008个gaps(补充数据表S4)。Illumina短读映射到组装的光皮梾木序列上,映射率为98.83%,覆盖率为99.44%。此外,约93.49%的mRNA测序读段映射到了基因组序列上。我们使用基准普适单拷贝直系同源基因(BUSCO)数据库评估了组装的完整性,显示97.71%的核心基因是完整的,组装的基因组序列中仅有0.9%缺失(补充数据表S5)。总体来看,这些结果表明组装具有高质量,准确性、连续性和基因组覆盖率均很高。
光皮梾木的基因组注释
重复序列
通过同源性和新颖方法识别了重复序列,从而确定了大约499.55 Mb(占55.22%)的重复序列(补充数据表S6)。长末端重复逆转座子(LTR-RTs)被发现是最丰富的转座元素(TEs),占基因组的48.83%,其次是DNA重复,仅占0.79%(补充数据表S7)。基于Kimura距离的拷贝发散分析揭示了没有检测到LTR-RTs和长间隔核内元素(LINEs)的显著近期积累(补充数据图S3),但在Ks峰值0.41处检测到一致的积累。基于与鹅掌楸相似的替换率(1.51 × 10^-9替换/位点/年)[22],我们估计这次TE爆发发生在大约135百万年前,使用公式TE爆发时间 = Ks /(2 × 核苷酸替换率)计算得出。LTRs被发现是这种古老重复拷贝数扩张的主要贡献者,增加了约1.2%,在41个单位(补充数据图S3),表明LTR-RTs在光皮梾木基因组扩张中发挥了关键作用。
基因识别与注释
在光皮梾木中,通过新颖、同源性和基于RNA-seq的预测组合进行基因识别和注释,预测出共31,640个编码蛋白的基因(补充数据图S4A和补充数据表S8),其中30,474个基因分布在11条染色体上(图1F)。染色体1上的编码蛋白基因数量最多(4943),而染色体5上有1702个编码蛋白基因(补充数据表S4)。在光皮梾木基因组中识别出共375个同源块,包括8152对基因对(图1F)。光皮梾木的平均编码序列长度为1158 bp,与调查的其他五种相关植物种类相似(补充数据图S4B),而平均基因长度为6739 bp更大(补充数据表S9)。预测的基因中,有29,852个(94.30%)基于同源性被赋予功能,这些同源性是与Swiss-Prot和NR数据库中注释的蛋白质相比较的,并且90.60%的基因包含了基于InterProScan功能注释的保守蛋白质域。超过一半(53.80%)的光皮梾木基因组中的基因成功地用基因本体(GO)术语注释,指示它们的功能类别。此外,京都基因和基因组百科全书(KEGG)途径数据库被用来将77.70%的基因映射到已知的植物生物途径(补充数据表S10)。共有21,783个基因同时在这些数据库中被注释(补充数据图S4C)。除了编码蛋白基因的注释外,还注释了ncRNA基因,包括880个miRNAs、727个tRNAs、676个rRNas和1335个snRNAs(补充数据表S11)。
比较基因组学分析
我们进行了涉及19种物种的比较基因组学分析,这些物种包括多个分类阶层,如山茱萸目(光皮梾木、C. controversa、喜树和珙桐),杜鹃花目(越桔、杜鹃花和猕猴桃),茄科星I(芝麻、番茄和咖啡),茄科星II(向日葵、莴苣和胡萝卜),蔷薇目(葡萄和月季),石竹目(石竹、甜菜和火龙果),以及无患子目(无患子)。这种广泛的物种选择被用来调查光皮梾木基因组的演化模式。分析了共39,759个基因家族,揭示了6,475个共有基因家族,以及254个光皮梾木基因组特有的基因家族(补充数据表S12)。对于每个物种,计算了正交群基因统计数据,光皮梾木拥有3,130个(10.27%)单拷贝基因和10,002个(32.82%)多拷贝基因(图2A和补充数据表S13)。对包括光皮梾木在内的选定植物物种的基因家族进行了簇分析,揭示了保守和特有的基因家族(补充数据图S5A)。识别出共720个特有基因(补充数据图S5B)。GO和KEGG富集被用来分析这些特有基因在光皮梾木中的功能。GO分析(调整后的P值<.05)显示光皮梾木中的特有基因涉及水解酶活性和碳水化合物代谢过程,表明它们在特定的催化水解反应中的潜在重要性(补充数据图S5C和补充数据表S14)。KEGG分析结果显示特有基因在氰氨酸代谢和萜类骨架生物合成中富集(调整后的P值<.05),表明它们可能参与疾病和害虫抵抗、免疫抑制和脂质降解的功能(补充数据图S5D和补充数据表S14)[23]。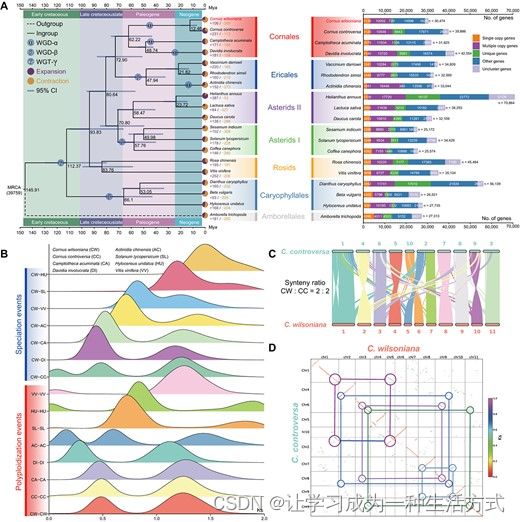
光皮梾木基因组的系统发育分析 A. 光皮梾木及其他18种植物的系统发育树。光皮梾木和其他已测序基因组的基因被分为五类,直接数字(n)显示在右侧的条形图上。基因家族的扩张或收缩用实心圆表示;总变化中相应的比例显示在饼图中。每个节点处标示的估计分化时间(百万年前,Mya)及其95%置信区间(CI)。嵌入文本的球体表示最近的WGD或WGT-γ事件。光皮梾木被突出显示。 B. Ks值揭示了光皮梾木演化过程中的两次WGD事件。 C. 光皮梾木与C. controversa之间的共线关系。背景中的所有带状线表示跨越>20基因的基因组间的同源块。 D. 光皮梾木与C. controversa之间的同源块点图。使用圆形和实线显示2:2的同源深度比例。
我们通过识别19种物种中的101个单拷贝基因家族进行了系统发育分析,结果显示光皮梾木与C. controversa有着密切的关系(补充资料图S6)。它们都是山茱萸科的成员,并在大约12.46百万年前分化(图2A)。随后,我们识别了269个收缩的和106个扩张的基因家族,分别包括1168和1105个基因(补充资料图S5B)。发现扩张的基因在与信号转导、ADP结合和信号接受器活性相关的GO项中富集(补充资料图S5E和补充资料表S15)。此外,我们对KEGG途径的砅究表明,扩张的基因家族在多个过程中显著富集,包括MAPK信号途径以及白藜芦醇、二芳基庚烷和姜酚的生物合成(补充资料图S5F和补充资料表S15)。这些发现表明,扩张的基因可能在植物对病原体攻击的反应中起了作用。此外,我们的砅究揭示了光皮梾木与Nyssaceae科的物种,特别是喜树和珙桐(图2A),在系统发育上的密切关系。Cornaceae与Nyssaceae的分化发生在大于62.22百万年前。这四个物种都属于山茱萸目。该目与杜鹃花目有姐妹关系,后者包括越桔、杜鹃花和猕猴桃(图2A)。这两个目之间的分化时间估计发生在大约72.90百万年前(图2A)。总体而言,基于19种物种的进化分析,我们为光皮梾木构建了一个相对全面且定义明确的物种系统发育路线。此外,与其他物种相比,我们表明光皮梾木在中新世期间经历了最终的分化,成为一个独特的物种。因此,光皮梾木在未来研究中作为晚期物种演化的潜在候选者具有前景。
为了深入了解物种分化和WGD事件,我们对光皮梾木和其他七种物种(C. controversa, Camptotheca acuminata, Davidia involucrata, Actinidia chinensis, Solanum lycopersicum, Hylocerus undatus和Vitis vinifera)进行了全基因组共线性分析。共识别出204,207对共线基因对和10,512个合成块(补充资料表S16)。通过分析同源和旁系基因对的Ks分布,推断出光皮梾木的WGD和物种形成事件,显示在约0.48和1.25处有峰值(图2B)。以前的砅究已经揭示了Vitis vinifera基因组中的三重同源基因块的特征模式,这归因于WGT-γ事件的发生[24]。这些三重复的基因组结构被认为起源于大约117百万年前发生的WGT-γ事件[25]。使用公式分化时间 = Ks/2r,我们可以推断光皮梾木在早白垩纪和古近纪时期分别经历了WGD事件,分别发生在大约44.90和115.86百万年前(补充资料表S16)。其中,古老的WGD事件(大约115.86百万年前)对应于WGT-γ事件(图2B),这是被子植物共有的[25]。此外,光皮梾木在与Nyssaceae科分化后不久(大约44.90百万年前)经历了更近期的WGD事件(图2A和B)。值得注意的是,这一近期的WGD事件也在其最近亲C. controversa中观察到,且在Nyssaceae科的喜树中也有类似的证据,这早于珙桐中WGD事件的发生(图2B)。因此,我们假设这一近期的WGD事件在山茱萸科内共享。除了三重事件外,我们还可以观察到与分化历史总体一致的显著物种形成事件(图2A和B)。然而,也有一些实例显示光皮梾木−Solanum lycopersicum(CW − SL,Ks = 1.19)和光皮梾木−Vitis vinifera(CW − VV,Ks = 0.82)的分化时间与山茱萸目−蔷薇目和山茱萸目−茄目的分化时间不一致,如图2A和B所示。我们将这种差异归因于使用单拷贝基因和共线基因对进行物种分化推断时引入的潜在偏差。因此,在特别砅究两个物种之间的分化时间时,采用共线基因对进行物种分化推断被认为是更可靠的方法。
在上述研究中,我们观察到光皮梾木和C. controversa之间存在密切的系统发育关系,并共享WGD和WGT-γ事件。结果显示,我们对这两个物种进行了更全面的全基因组共线性分析,揭示了在全基因组水平上显著的染色体共线性(图2C)。通过分析它们的同源基因,可以清晰地观察到2:2的同源比例(图2D)。通过比较形成三个Ks峰的同源基因块,注意到最近的峰(Ks = 0.09)包含的共线基因对数量最少(图2B)。这表明它们在最终分化期间保留了广泛的相似染色体特征,这与图2C中显示的大型共线片段一致。
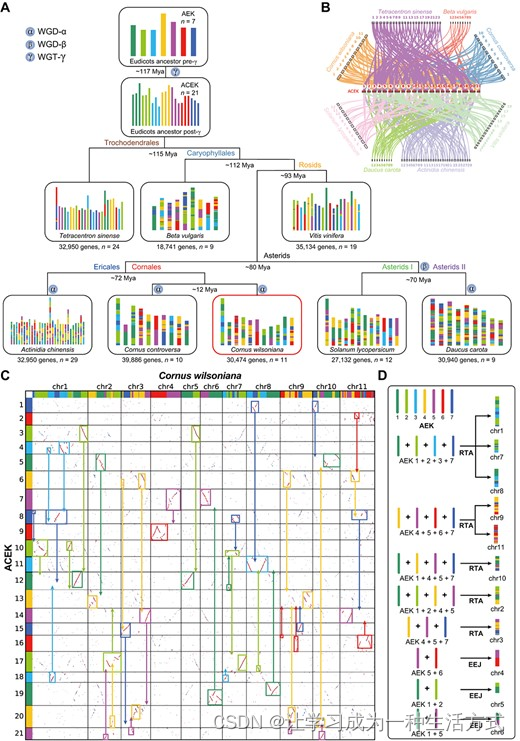
光皮梾木的进化情景 A. 底部描绘的现代基因组,不同的颜色反映了起源于7个祖先染色体(n=7 AEK)的起源(顶部)。 B. ACEK与八个特定物种之间的共线关系。 C. 光皮梾木与ACEK之间的同源块点图。染色体的重组通过矩形和实心箭头显示。 D. 光皮梾木染色体起源的示意图。
光皮梾木的核型演化
核型演化代表了从祖先基因组到现代基因组的染色体变化。这一信息对于重建祖先核型和追踪现代物种的逐步演化至关重要。为了评估光皮梾木的古历叚,我们对八个物种(Tetracentron sinense,Beta vulgaris,Vitis vinifera,Actinidia chinensis,C. controversa,光皮梾木,Solanum lycopersicum,Daucus carota)的核型演化进行了分析。我们用Tetracentron sinense重建了被子植物祖先核型(AEK),它是被子植物早期分化系之一。在WGT-γ事件之后,AEK演化为祖核被子植物核型(ACEK),具有21个原始染色体(图3A)。随后,核心被子植物物种通过这21个原始染色体的融合和裂解事件,产生了不同的染色体数目和组成。据此,我们识别了ACEK和八个特定物种之间共享的同源基因块,以评估物种的核型(图3B)。
接着,我们根据八个物种的系统发育关系和同源基因块重建了代表性物种的核型演化轨迹(图3A)。从这一分析中可以明显看出,Tetracentron sinense和Vitis vinifera保留了相对完整的原始染色体。随着物种的多样化和经历了WGD事件,光皮梾木的原始染色体组成变得异常复杂。尽管如此,我们仍然可以推断光皮梾木每个染色体内不同片段的起源(图3C)。此外,ACEK和光皮梾木之间明显存在1:2的同源性比例,再次确认了光皮梾木在WGT-γ之后发生了另一次WGD事件(图3C)。
鉴于光皮梾木的巨大分化程度,详细描述其核型演化历史成为一项复杂的任务。然而,通过建立光皮梾木和AEK之间的连接,通过与ACEK的同源基因块以及采用相互转位的染色体臂(RTAs)、嵌套染色体融合(NCF)和端-端连接(EEJ)等机制,我们可以推断染色体的起源(图3D)。例如,chr1,chr7和chr8被认为是通过RTA从AEK的第1、2、3和7原始染色体形成的,而chr9和chr11则通过涉及AEK的第4、5、6和7原始染色体的RTA形成。此外,chr4,chr5和chr6是通过两个不同的AEK原始染色体通过EEJ融合的结果(图3D)。我们相信,随着物种基因组数量的不断增加,未来的研究努力将使我们能够揭示核型演化的复杂细节。
识别参与油脂生物合成的关键基因
为了发现负责光皮梾木油脂生物合成的关键基因,进行了比较基因组分析。结果显示,光皮梾木拥有1663个参与酰基脂肪酸合成的基因,与大豆(1695个基因)相当,但高于非油籽植物如拟南芥(1290个基因)。与拟南芥相比,光皮梾木和大豆在脂肪酸基因功能组中显著富集(补充数据表S17)。尽管在某些类别的脂肪合成基因中基因数量有轻微变化,但光皮梾木在质体脂肪酸合成、脂质信号传导和三酰甘油(TAG)合成中具有更多的基因参与。
为了探索与脂肪酸和TAG生物合成相关的基因,我们手动检查了光皮梾木的蛋白质组并将其与UniProt参考簇(UniRef)数据库进行了比对,从而识别出80个候选基因。其中55个通过RNA测序数据显示出花蕾、花和叶组织中的表达信号(补充数据表S18)。这些基因构成了一个完整的油脂生物合成途径,丰富了脂肪酸生物合成(ko00061)的KEGG途径(图4A和B)。理解这些代谢基因的表达多样性对于提高油品质参数至关重要,特别是在实现O/L比率方面。FAD2,编码δ-12油酸脱氢酶并控制高油酸性状[28],在盛花期表达高,但种子脱水后表达低(图4A)。参与TAG途径的关键酶在不同发育阶段的表达水平呈现变化(图4A)。与其他作物已知的FAD蛋白基因相比,发现光皮梾木的FAD蛋白与向日葵[29]、核桃[30]和油茶[31](这些作物不饱和酸比例高)的FAD蛋白具有密切的进化关系(图4C)。这些FAD蛋白包含两个相同的域(PF00487和PF11960)(补充数据图S7),表明在调节O/L比率方面具有类似的功能。此外,在参与TAG生物合成的55个基因中,CW06G17070基因在高产和低产果实样本之间的基因表达存在显著差异[log2(倍数变化)=2.85,调整后的P值=1.46e−6](图4D和补充数据表S19)。
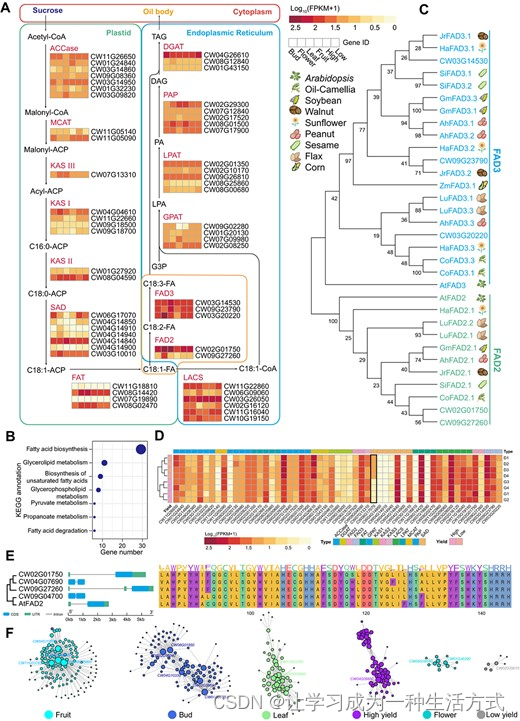
涉及油脂生物合成的基因 A. 油脂生物合成途径的简化表示。通过测序花蕾、花朵、果实和叶子的 RNA 样本,为每个基因估算的表达水平,通过 FPKM 表示,用于高产和低产基因型。 B. 光皮梾木 油脂生物合成基因的 KEGG 途径注释。 C. 10 种物种中编码 FAD2 和 FAD3 的候选基因的系统发育树。分支旁的值表示该分支的最大似然估计 (MLE) 引导值。系统发生树右侧的卡通插图显示物种类型。 D. 高产和低产基因型果实中假定的油脂生物合成基因表达水平的热图。 E. 光皮梾木 和 AtFAD2 中四个同源 FAD2 基因的 DNA 结构和蛋白质域 PF00487 的比较分析。 F. 基于 WGCNA 构建的共表达调控网络。节点大小代表连接度,转录因子在节点周围标记。
根据先前的研究,拟南芥中只鉴定出一个 FAD2 基因(登录号 L26296)[32]。然而,在其他油料作物中至少鉴定出了一个额外的基因。例如,在大豆(登录号 L43920 和 L43921)和橄榄(登录号 AY733076 和 AY733077)中鉴定出两个 FAD2 基因[33, 34],在向日葵(登录号 AF251842、AF251843 和 AF251844)和油菜籽(登录号 HQ008320、HQ008321 和 HQ008322)中鉴定出三个基因[35, 36],在芸薹属植物中鉴定出四到六个不同的 FAD2 基因[37]。同样,在 光皮梾木 中,使用 AtFAD2 基因(AT3G12120)作为查询,鉴定出四个表达显著的同源 FAD2 基因(同一性>70%,e值<1e−5,FPKM>1)。多个 FAD2 基因的存在可能表明其在调节 O/L 比率方面的潜在协调作用。此外,基于 DNA 序列分析(图 4E),CW02G01750 和 CW09G27260 与已知的 AtFAD2 基因共享类似的基因结构特征,包括 5′-非翻译区(UTR)的单个大内含子和单个编码序列。另一方面,CW09G04700 和 CW04G07690 缺少 UTR 区域,但具有与 AtFAD2 相似长度的编码序列。在蛋白质序列结构方面,PF00487 域与脂肪酸脱饱和酶相关,光皮梾木 中的四个 FAD2 蛋白的氨基酸组成与已知 AtFAD2 中的 PF00487 域高度相似(图 4E)。此外,我们观察到在每个位置上氨基酸类型的相对保守性(图 4E),为未来的基因工程努力奠定了基础。最后,为了全面评估参与油脂生物合成的关键基因,我们采用加权相关网络分析(WGCNA)识别了果实中的四个枢纽转录因子(CW02G05600、CW04G10500、CW08G0580、CW11G03760)(图 4F 和补充数据图 S8)。我们还鉴定了与这些转录因子共表达的下游基因,P 值 <1e−5,GS > 0.9(补充数据表 S20)。我们相信这些关键基因将为 光皮梾木 中 TAG 生物合成相关的育种研究提供基本依据。
花蕾发育中的转录和代谢
花蕾发育过程中的转录和代谢是植物开花的关键过程[38]。在这里,我们采用转录组学和代谢组学技术研究花蕾发育过程中的变化。三个生物重复的主成分分析(PCA)展示了高数据可重复性(补充数据图 S9 和 S10)。在花蕾的各个发育阶段,即花萼分化阶段(H1 和 L1)、雌蕊分化阶段(H2 和 L2)和雄蕊分化阶段(H3 和 L3),共有 2721 个基因和 424 个代谢物显示出显著的差异表达(图 5A,补充数据表 S21 和 S22)。有趣的是,一些最显著的差异表达基因(DEGs)和差异表达代谢物(DEMs)在同一发育阶段的高产(H)和低产(L)性状中被检测到,例如在 H1 对 H2 和 L1 对 L2 比较中的 CW09G17280,以及在 H2 对 H3 和 L2 对 L3 比较中的 CW11G04360 和 lysoPC(20:3),表明它们可能对花形态产生影响。这些 DEGs 和 DEMs 还认为参与了相同的 KEGG 途径,如苯丙素生物合成和黄酮生物合成(图 5B),这与 光皮梾木 的颜色和香气形成相关[39]。为了进一步研究代谢物和基因之间的相关性,我们进行了 DEGs 和 DEMs 的趋势分析。我们鉴定了共 10 个簇(图 5C,补充数据图 S11 和补充数据表 S23),这些簇集体展示了在高产和低产性状中 DEGs 和 DEMs 一致的变化模式。此外,在这 10 个簇中,我们对富集在苯丙素生物合成途径(ko00940)和黄酮生物合成途径(ko00941)中的 DEGs 和 DEMs 进行了相关性分析。这种分析揭示了几对强相关的基因-代谢物对,如簇 2 中的 CW05G15020-咖啡酰齐墩果酸、簇 4 中的 CW01G00140-松木素和簇 9 中的 CW08G05520-苹果素(补充数据图 S12)。
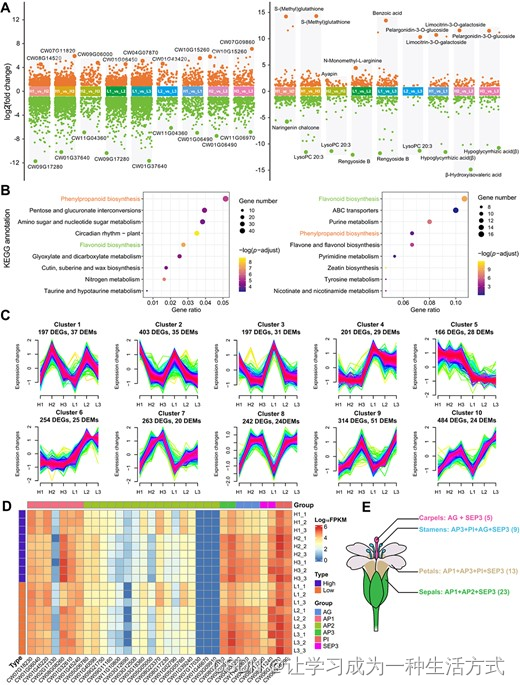
涉及花蕾发育的基因和代谢物 A. 不同表达基因(DEGs,左图)和不同表达代谢物(DEMs,右图)的散点图。点表示上调或下调的基因和代谢物。每个条形图上突出显示了差异最显著的基因和代谢物。 B. 光皮梾木 假定的花蕾发育基因和代谢物的 KEGG 途径注释。调整后的 P 值 <0.05。 C. 趋势分析识别了 10 种不同的 DEG 和 DEM 表达模式。x 轴代表高产和低产性状的三个发育阶段,而 y 轴代表各阶段 log2 转换后的归一化强度比。 D. 花蕾发育中 MADS-TF 基因表达的热图。三个发育阶段的花蕾,即花萼分化阶段(H1 和 L1)、雌蕊分化阶段(H2 和 L2)和雄蕊分化阶段(H3 和 L3),来自两种基因型:高产(H)和低产(L)。 E. 花器官的 MADS-TF 基因共调控景观。括号中显示 MADS-TF 基因的数量。
此外,花蕾分化不仅是组织发育和功能提升的过程,还是营养积累的过程。可溶性营养物,如蔗糖和氨基酸,从营养器官运输到花蕾,随后转化为不溶性聚合物化合物,如淀粉、蛋白质和脂肪酸。这一渐进过程最终形成了花的主要成分,并为其进一步开花做准备。在开花阶段,各种植物器官,特别是花器官,含有大量的 MADS-TF 基因。在 光皮梾木 的情况下,我们识别了 33 个 MADS-TF 基因(补充数据表 S24),其中包括六个关键转录因子,AP1(APETALA1)、AP2、AP3、AG(AGAMOUS)、PI(PISTILLATA)和 SEP3(SEPALLATA3),直接与花的发育相关。在这些中,除了 AP2 组的 CW07G17030、CW01G46670 和 CW03G09910 外,所有转录因子在花的发育期间表达水平显著。先前的研究表明,MADS-TF 基因协同作用以构成花器官的多样形态。因此,我们重建了控制 光皮梾木 花萼、花瓣、雄蕊和心皮发育的转录因子共调控景观(图 5E)。总体而言,这些发现为花蕾的发育提供了宝贵的见解,并为 光皮梾木 的分子育种提供了遗传资源。
讨论
通过生成 光皮梾木 的草图基因组,我们的研究为理解山茱萸科内油脂生物合成和花蕾发育的遗传基础和演化提供了宝贵资源。在本研究中,我们使用 PacBio 和 Hi-C 数据生成了 光皮梾木 基因组的染色体级组装。系统发育分析确定 C. controversa 为与 光皮梾木 最接近的物种,共同从山茱萸科分化出来,大约在 12.46 百万年前。值得注意的是,光皮梾木 与鼠刺科物种关系密切。我们的研究概述了各种物种之间的分化时间,为 光皮梾木 构建了明确的物种系统发育路线,并表明其具有研究晚期物种演化的潜力。通过 Ks 分布分析推断出 光皮梾木 的 WGD 和物种形成事件,表明古老和近期的 WGD 事件分别发生在大约 115.86 百万年和 44.90 百万年前。这一近期的 WGD 事件在山茱萸科内共享,并在 光皮梾木 及其最近亲缘物种的分化中发挥作用。我们采用 RTA、NCF 和 EEJ 等机制推断染色体的起源,这揭示了 光皮梾木 染色体型复杂的进化历史。
由于其强大的降脂效果,光皮梾木 果实的高油脂含量近年来越来越受到关注。在本研究中,我们识别了参与 光皮梾木 油脂生物合成的关键基因和途径。我们的结果表明,光皮梾木 与其他高油作物共享常见途径,如棕榈酸、油酸和亚油酸的生物合成。然而,我们还发现了可能有助于 光皮梾木 果实高 O/L 比率的潜在基因,如 FAD2 基因家族成员。这些发现为进一步研究 光皮梾木 及其他高油作物油脂生物合成的遗传基础和药用价值奠定了基础。此外,我们还研究了 光皮梾木 花蕾发育的遗传基础。我们的分析识别了几个参与花蕾发育的关键 MADS-TFs,包括 AP1 和 SEP 同源基因。我们发现这些基因在不同植物物种中高度保守,对花器官的发育至关重要。通过我们的研究获得了关于 光皮梾木 花蕾发育分子机制的见解,这可能为培育具有优越花卉特征的新品种铺平道路。
总体而言,我们的研究为理解 光皮梾木 的演化和关键性状的遗传基础提供了宝贵资源。光皮梾木 基因组的染色体级组装可以促进与重要性状相关的基因和途径的发现,并可能加速具有更好花卉特征和油脂含量的新品种的培育。此外,我们的研究对植物漜化和其他植物物种复杂性状的遗传机制也有一定的影响。进一步的功能研究关键基因是必要的,以加深我们对 光皮梾木 及其他高油含量椗物的关键性状遗传基础的理解。
材料和方法
植物材料
本研究中使用的生物样本来自 光皮梾木 组织材料,这些材料是从广东省林业科学院种源银行的种植田(113°38′0″, 23°20′0″)采购的。研究包括了不同发育阶段的花蕾:花萼分化阶段(H1 和 L1)、雌蕊分化阶段(H2 和 L2)以及雄蕊分化阶段(H3 和 L3)。这些阶段在两种基因型中进行了研究,这两种基因型分别以高产量(H)和低产量(L)为特征。每个样本共收集了三个独立的生物重复。此外,花样本在初始开花阶段(F1)和盛花期(F2)独立采收,每个阶段两个生物重复。在初始开花阶段之前收集了新鲜嫩叶(Le),代表两个生物重复。从两种不同基因型的种子中收获,这些基因型分别标记为高产量(G)和低产量(D),采收是在脱水后进行的。每个样本收集了四个生物重复。所有收集的材料都使用液氮迅速冻结,并存放在 -80°C 下,直到进行 RNA 测序和代谢物分析。
DNA提取和测序
为了从光皮梾木的新鲜嫩叶中提取DNA,采用了十六烷基三甲基溴化铵(CTAB)方法,因为其能够提供高质量基因组DNA。随后,所获得的DNA进行了片段化处理,并用于创建插入片段>30 kb的文库。我们的具体策略是选择插入片段为20 kb的DNA片段,随后在PacBio Sequel平台上进行测序。总共使用了六个SMRT细胞进行测序过程。原始PacBio子读数通过SMRT Link流水线处理,得到了约6.12百万子读数。这些子读数的平均长度为22.9 kb,实现了150×的基因组覆盖率,生成了总共120 Gb的测序输出。为了处理可能的组装错误,使用同一DNA样本准备了Illumina DNA-seq文库。在Illumina HiSeq X Ten平台上进行了350-bp读数的配对末端测序,得到了116 Gb的数据集和120×的基因组覆盖率。对于Hi-C实验,将约3 g的新鲜嫩叶组织在液氮下粉碎成细粉。这些材料经历了一系列步骤,包括染色质提取、酶解、DNA连接、纯化和片段化,最终生成了Hi-C文库。这个精心准备的文库使用Illumina HiSeq X Ten平台进行了测序,显著地促进了染色体级基因组组装的实现。有关数据集的详细信息可以在补充数据表S2中找到。
基因组大小估计
为了确定光皮梾木的基因组大小,我们采用了传统的k-mer计数技术。这涉及使用Jellyfish [46]分析清洁的Illumina短读数(大小为17 bp)中的k-mer出现情况。我们的测序深度评估依赖于识别k-mer出现频率分布曲线中的峰值,特别关注同源峰特征。通过分析总共89,452,735,820个k-mer,我们确定了对应于同源峰的k-mer深度,发现为94。考虑到k-mer深度为1可能由错误引起,我们通过考虑1.08%的错误率来进行了处理。然后使用这个错误率来完善我们的基因组大小计算。基因组大小的初步估计是根据总k-mer计数与同源峰深度的比率得出的。为了进一步提高准确性,我们纳入了错误率,得到了修正后的估计:(k-mer计数)/(同源峰深度)×(1 − 错误率)。基于这些计算,光皮梾木的估计基因组大小被确定为约951.62 Mb。然而,在应用校正后,修正的基因组大小被重新计算为约941.39 Mb(如补充数据表S3中详细描述)。此外,使用GCE v1.0.2 [47]来估计杂合比例和重复序列的比例。
基因组去新生装配
对于去新生装配,我们使用Falcon v2.0结合PacBio清洁读数[48]。装配过程中包含了特定参数seed_coverage=30和length_cutoff_pr=4 K,这有助于获得最佳结果。随后,初步草图装配经历了精炼步骤。使用PacBio清洁读数,应用Arrow v2.2来打磨装配[49]。在此之后,使用Illumina清洁读数执行了进一步的打磨,使用的是Pilon v1.22[50]。为了消除任何冗余的连锁群并增强装配的简洁性,我们将Purge_Haplotigs纳入我们的流程[51]。确保装配的完整性和均匀的测序覆盖率是关键。为此,我们使用Minimap2 v2.5的默认参数,对最终装配进行了连续长读数的重新比对[52]。在我们评估组装基因组的质量的追求中,我们进行了全面的分析。我们使用了BUSCO v3.0.2[53],利用Embryophyta_odb10数据库,并遵循默认参数。这种评估使我们能够衡量装配基因组中基因内容的完整性。
染色体级装配使用Hi-C
为了整合Hi-C数据提供的结构信息,我们执行了一系列步骤来精炼我们的支架装配。最初,Hi-C清洁数据使用BWA-mem v0.7.17[54]与支架装配进行了比对。我们采用了严格的选择标准,特别是利用读数对在约束位点附近500 bp内的连锁群进行比对的读对。此外,只考虑了独特映射的读对,代表有效的互动配对末端读取。这些精心挑选的读对随后被用来构建伪染色体序列,提高了我们基因组表示的准确性。使用LACHESIS结合Juicebox v1.8.8[55, 56]进行了支架的组装、排序和定向,以形成光皮梾木特有的11条染色体的连贯结构。为了直观地展示Hi-C数据中的互动模式,我们生成了Hi-C矩阵热图。这个热图,提供了染色体互动的洞察,是使用HiCExplorer 3.7.2[57]创建的,分辨率为500 kb。
基因组注释
我们采用双重方法进行重复注释,结合同源比对和去新建搜索技术,全面识别全基因组重复序列。我们使用 TRF 工具通过原位预测检测串联重复序列。为确保全面评估,我们利用了公认的同源预测资源 Repbase 数据库。使用 RepeatMasker 软件及其内部脚本(RepeatProteinMask),默认参数帮助提取重复区域。我们使用 LTR_FINDER、RepeatScout 和 RepeatModeler 构建去新建重复元素数据库。在构建原始转座元件库时,我们倾向选择长度超过 100 bp 且含有小于 5% 'N' 核苷酸的重复序列。我们合并 Repbase 和去新建转座元件库,创建了定制的复合库,随后经过 uclust 策划处理,形成了非冗余库。这个精炼库随后被提供给 RepeatMasker 进行全面的重复注释。
结构注释
基因组中基因模型的全面注释采用了包括原位预测、基于同源性的预测和 RNA-seq 辅助的方法。为了提高同源预测的准确性,我们从 NCBI 获得了一组经过策划的同源蛋白序列。使用 TblastN v2.2.26,这些蛋白序列与基因组比对,建立序列同源性。使用 GeneWise v2.4.1 工具优化此对齐并解析识别蛋白区域内的基因结构。原位基因预测过程包括多种工具的使用,共同预测重复屏蔽基因组内的编码基因。将 RNA-seq 数据从幼叶对齐到 光皮梾木 基因组,这一过程为基因组基础的转录体组装奠定了基础。使用 Cufflinks 组装后,我们使用 EvidenceModeler 合并通过三种方法预测的基因,生成简洁的参考基因集。最后,特定基因家族的选择由领域专家进行手动策划,确保了基因注释结果的精确性。
功能注释
为了全面指定基因功能,我们采用了多方面的方法,整合了序列比对、基序/域注释和本体赋值。首先,蛋白序列精确地对齐到 Swiss-Prot 数据库。通过使用 InterProScan,我们详尽地探索了广泛的公共数据库,识别了一系列潜在的基序和域。将 GO ID 精确赋值,这些赋值基于 InterPro 数据库中的相应条目。我们还利用 KEGG 途径数据库,将基因集映射到相关途径,从而增强了我们对每个基因运作功能环境的理解。
非编码 RNA 注释
我们使用专门的 tRNAscan-SE 程序完成 tRNA 的预测。采用比较方法预测 rRNA,使用与近亲种的 rRNA 序列作为参考模板。为了全面识别其他非编码 RNA 类,我们使用 Infernal 软件系统地识别这些功能显著的 RNA 分子。
种间基因组比较分析
基因家族和系统发育分析
为了全面了解 光皮梾木 基因组中存在的蛋白编码序列,我们进行了涵盖 18 种不同物种的比较基因组学研究,这些物种跨越了多个分类阶。这些物种包括 Cornales(C. controversa [85]、Camptotheca acuminata [86] 和 Davidia involucrata [87])、Ericales(Vaccinium darrowii [88]、Rhododendron simsii [89] 和 Actinidia chinensis [90])、asterids I(Sesamum indicum [5]、Solanum lycopersicum [91] 和 Coffea canephora [92])、asterids II(Helianthus annuus [6]、Lactuca sativa [93] 和 Daucus carota [94])、rosids(Vitis vinifera [95] 和 Rosa chinensis [96])、Caryophyllales(Dianthus caryophyllus [97]、Beta vulgaris [98] 和 Hylocereus undatus [99])和 Amborellales(Amborella trichopoda [100])。为了表示具有选择性剪接变异的基因,我们选择了包含最长编码序列的转录本作为每个基因的代表。我们使用 BLASTp 进行序列对比,设定严格的 e 值截止值为 1e-5,以便深入比较。OrthoMCL v2.0.9 工具箱在我们探索基因家族成员资格时证明无价。采用全面的方法,我们进行了全对全的蛋白序列相似性搜索,并使用马尔科夫链聚类来识别复杂的关系。从 OrthoMCL 的结果中,提取了 101 个单拷贝同源基因。接着,我们使用 Muscle v3.8.31 和默认参数创建了一个序列对齐,使用 Gblocks v0.91b 来精确消除对齐中的歧义位置。
为了解开进化的复杂分支,我们使用 RAxML v8.2.12 构建系统发育树。使用 Amborella trichopoda 的数据集作为这一分析的外群。通过执行 1000 次自助分析进行稳健性评估。对进化的时间方面感兴趣,我们采用了 MCMCTree 中的贝叶斯松弛分子钟方法来估计物种分化时间。我们依赖 TimeTree 数据库的校准时间,特别是围绕 光皮梾木 和 Amborella trichopoda 之间的分化。为了阐明基因家族动态,我们采用了 CAFE v5.0.0,将基因家族演化概念化为随机的出生和死亡过程。此分析指定了参数 -p 0.05 -r 1000。此外,我们深入探讨了基因家族演化的复杂细节,这涉及到在系统发育树中基因家族大小变化的详细分析。为了辨别功能影响,我们对扩大的基因家族进行了 GO 和 KEGG 富集分析,这些分析是通过 clusterProfiler 4.0 进行的。
全基因组复制分析与识别
为了识别全基因组复制(WGD),我们使用了 DIAMOND v0.9.29.130 [73] 来精确地定位显示出显著序列相似性的基因对(e值小于1e−5)。为了确保我们发现的稳健性,我们采用了严格的 C 分数阈值。具体来说,保留了 C 分数大于0.5的基因对,这一过滤过程由 JCVI 软件 [109] 促成。为了揭示这些相似基因对的空间关系,我们采用了 MCScanX [110],这是一个专门设计来辨识染色体上的邻接模式并建立共线基因块的强大工具。此分析的结果让我们能够识别出保守的基因组组织实例,揭示了复制基因块的进化轨迹。共线性模式通过共线性图表形象地展示,这些图表是用 JCVI 软件 [109] 精心绘制的。为了深入探讨 光皮梾木 基因组内 WGD 事件的复杂性,我们转向使用 wgd v1.1.0 软件 [111]。这个为 WGD 分析量身定做的专用工具帮助识别了大规模复制事件塑造基因组景观的实例。为了深入了解这些 W G D 事件的进化意义,我们使用了 PAML 中的 codeml 工具 [105]。这种多方面的方法使我们能够更深入地理解 WGD 对 光皮梾木 基因进化的影响。
核型演化
这一全面的分析套件通过使用 WGDI [112]——一个在解析基因组数据中的复杂模式中不可或缺的工具——来进行协调。这包括了一系列系统的步骤,每一步都有助于对基因组组织和演化的整体理解。为了开始探索 Tetracentron sinense 的染色体结构,我们结合使用了 WGDI 和 -d 参数。这种方法促成了点图的创建,使我们能够直观地辨识出潜在的染色体结构。随后,我们调用 WGDI 的 -ak 参数来揭示由 Tetracentron sinense 推断出的 AEK 和 ACEK 组件。这些推断元素为祖先基因组的组织提供了关键洞见。对于其他正在考虑的物种,我们深入探讨了基因组间关系的领域。利用 -icl 参数,我们发掘了 ACEK 和特定物种之间的共线基因,提供了基因组组织的比较视角。随后,我们利用 -bi 参数识别了这些共线块之间的共线基因。为了简化分析并促进共线片段的识别,我们谨慎地使用 -bi 参数整合了这些共线块。这种整合使我们能够精确定位代表共享进化历史的连贯基因组区域。分析继续进行了严格的过滤过程。使用 -c 参数,我们系统地排除了可能源自更古老进化事件的块,确保关注点保持在相关的进化模式上。为了验证剩余块的合理性,我们采用了 -bk 参数。这一关键的验证步骤确保了正在考虑的共线块展示了一致的进化轨迹。对于 AEK 的映射,我们使用了 -km 参数,最终阐明了 AEK 在不同物种间的复杂映射。为了形象地概括种间的共线性,我们使用了 JCVI 软件 [109],生成了种间共线性图。这些图表将复杂的基因组关系转化为连贯的视觉图像,提供了共享进化动态的快照。
参与油脂生物合成的基因及其分析
为了阐明控制油脂生物合成的遗传组成部分,我们启动了一项对相关基因的详尽检索和比较分析。这项分析的基础在于全面的 aralip 数据库(Acyl Lipids: downloads),该数据库收录了与拟南芥油脂生物合成相关的基因。为了发现大豆和 光皮梾木 中的同源基因,我们进行了序列相似性搜索。使用严格的标准(e值 ≤1e−5,对齐同一性 ≥40%,对齐覆盖率 ≥50%)部署 DIAMOND 工具,我们建立了基因之间的联系。这些连接随后被映射回 aralip 数据库中的类别,最终实现了与油脂生物合成相关的基因的量化。同时进行了一项平行调查,涉及搜索 80 个与 光皮梾木 油脂生物合成相关的基因。这次搜索针对的是 UniProt 参考簇(UniRef)数据库(UniProt)使用 光皮梾木 的蛋白质组进行。与前一次分析相同参数的 DIAMOND 工具,使我们能够识别同源基因。这些同源基因进一步检查表达信号,其中 55 个基因在至少一个样本中显示信号。利用这些基因,我们开始设计 TAG 生物合成途径。
为了理解脂肪酸脱饱和酶(FAD)蛋白的进化关系,我们设计了一种复杂的策略。从检索八种含油作物(玉米、亚麻、芝麻、花生、向日葵、核桃、大豆和油茶)和拟南芥中的 FAD 蛋白开始,我们以 光皮梾木 的 FAD 蛋白为基础。这个数据集用于通过最大似然方法构建系统发育树,通过 MEGA 11 [113] 实现。通过计算 1000 个自助复制,增强了分析的准确性,同时将小于 50% 的值谨慎排除。通过从 InterPro(InterPro)检索蛋白质结构域来详细说明 FAD 蛋白的结构特征。GeneDoc 2.7(GeneDoc HomePage)促进了基因结构的全面可视化。为了深入理解结构域核苷酸组成,使用了 ggmsa 工具 [114]。在基因共表达网络领域,我们利用了 R 包 WGCNA [115] 的功能。这个工具在构建揭示基因相互作用的复杂网络方面至关重要。为了可视化这些网络,无缝集成了 igraph(igraph – Network analysis software),确保了复杂关系的连贯表现。
转录分析
样本采集后使用 pBIzol 试剂盒(BIOFLUX,杭州博瑞科技,中国杭州)进行 RNA 分离和纯化。随后,构建了 cDNA 文库并进行了测序。提取 RNA 样本的质量评估是流程中的关键步骤。这是通过一系列技术实现的。NanoDrop 紫外分光光度计(Thermo,沃尔瑟姆,马萨诸塞州,美国)帮助 RNA 定量,而样本的完整性则通过 Bioanalyzer 2100 系统(Agilent,圣克拉拉,加州,美国)进行评估。这一全面评价确保了 RNA 适用于下游分析。在构建 cDNA 文库时,我们整合了约 3 微克原始 RNA 样本。这些精心准备的文库为后续测序工作奠定了基础。利用 Illumina HiSeq 4000 平台的能力,我们开始了高通量测序,生成了跨度为 150 bp 的配对末端读取。这一测序工作由奥韦基因生物技术有限公司(中国北京)的专家负责。原始测序读取数据以 FASTQ 格式存在,并经过严格的预处理。使用 Trimmomatic v0.39 精确去除接头和低质量序列。这种以质量为中心的处理确保了下游分析将基于高保真序列。为了与 光皮梾木 的参考基因组建立复杂的比对,我们使用了 STAR v2.7.10b 工具。这一比对过程促进了处理读取的准确映射,为基因表达模式的详细探索铺平了道路。为了量化基因表达水平,我们转向使用 RSEM 1.3.3,这是一个在计算 FPKM 方面闻名的精确工具。这一指标作为基因表达动态的强有力指示器。为了识别差异表达基因,这是我们分析的关键方面,我们部署了 DESeq2 v1.34。这个工具专注于统计显著性和生物学相关性,帮助区分表达模式发生变化的基因。我们的差异表达标准包括 P 值阈值 ≤0.05 和绝对 log2(倍数变化) 值 >1。
代谢组分析
使用花蕾样本进行代谢组分析,样本来自花萼分化阶段、雌蕊分化阶段和雄蕊分化阶段。在进行超高效液相色谱-质谱(MS/MS)分析之前,样本首先被冻干然后研磨成细粉。质谱数据使用 Analyst 1.6.3 处理。通过 MS 使用本地代谢数据库对样本中的代谢物进行了识别以及定量和定性分析。为了进行主成分分析(PCA)和偏最小二乘判别分析(PLS-DA),我们使用了 ropls v1.26.4 包来识别显著变化的代谢物,阈值设定为 |log2(倍数变化)| ≥1,VIP ≥1,且 P 值 ≤0.05。


















 4007
4007

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









