








题目:AlphaFold2 and Deep Learning for Elucidating Enzyme Conformational Flexibility and Its Application for Design
文献来源:https://doi.org/10.1021/jacsau.3c00188 (JACS)
代码:无 综述
内容:最近,AlphaFold2(AF2)和其他深度学习(DL)工具在准确预测蛋白质和酶的折叠三维(3D)结构方面的成功,使结构生物学和蛋白质设计领域发生了革命性的变化。三维结构确实揭示了酶催化机制排列的关键信息,以及哪些结构元素控制了活性位点口袋。然而,理解酶的活性需要详细了解催化循环中所涉及的化学步骤,并探索酶在溶液中所采用的多种热可及构象。在这一观点中,最近的一些研究说明了AF2在阐明酶的构象景观方面的潜力。本文讨论了基于af2和DL的蛋白质设计方法的关键发展实例,以及一些酶设计案例。这些研究表明了AF2和DL在允许高效酶的常规计算设计方面上的潜力。
1.研究背景
Alphafold2 (AF2)解决了一个存在了60年的问题:通过一级序列来了解蛋白质/酶的3D结构。AF2是一种深度学习(DL)算法,它结合了基于蛋白质结构的进化、物理和几何约束的神经网络结构,能够高精度地预测蛋白质的三维结构。AF2被认为是蛋白质结构预测的里程碑之一,并促进了DL方法在许多其他场景中的应用。虽然AF2算法在预测酶的天然能量最低结构方面具有非常好的性能,但是了解单一的静态折叠结构并不足以理解和实现工程功能。此外,这些方法的另一个局限性是在于非蛋白质部分(即辅助因子、底物、金属离子)没有被预测。
酶的三维结构确实能提供关于催化机制和结构元件的排列的非常相关的信息,但了解酶的功能需要探索酶在溶液中采用的热可及构象的集合。这种构象的集合可以在所谓的自由能景观(FEL,见图1中不同反应阶段的FEL)中表示-显示热可及构象的相对稳定性以及分离它们的动力学势垒。构象的改变会直接影响催化功能,包括快速时间尺度上的侧链构象变化,环运动在较慢的时间尺度上通常在底物结合/产物释放中起关键作用,在某些情况下通常对应于最慢的变构转变过程。对自然酶和进化酶的构象景观的评价提供了相关的新见解。实验x射线结构和相关的b因子,室温x射线实验和核磁共振实验已经被用来探索由实验定向进化技术产生的几种酶变体的突变引起的构象景观的变化。从计算的角度来看,FEL的重建以及突变后如何发生的变化为理解酶的功能(以及设计)提供了关键信息。

图1 模型酶的催化循环和自由能景观(FEL)在不同步骤中的相关构象变化:自由酶(E)、酶−底物(ES)和酶−产物(EP)。对于FEL重建,需要定义一些关键的自由度(DOF)。
最近,不同的研究小组表明,AF2实际上可以提供同一蛋白质的多种构象,这表明AF2在阐明酶和蛋白质的构象景观方面具有潜力。考虑到AF2的计算成本相当低,特别是与分子动力学(MD)模拟计算要求相比,其应用于评估突变对构象景观的影响是非常有吸引力的。这可能会影响基于AF2的构象聚焦酶设计协议的发展。
2.构象动力学在酶的功能和进化中的作用
酶具有高度预先组织的催化残基完美地排列的活性位点口袋,可以有效地稳定一个特定反应的过渡态(s)。这种预组织是由酶的能力采取多种构象重要的底物结合和/或产物形成的。构象灵活性的重要性清楚地显示出催化抗体的设计,呈现一个理想的互补结构-显示出相对于酶明显较低的催化活性。这表明高效催化不仅需要过渡态稳定,还需要构象集合的优化。事实上,酶通过自然或实验室进化来适应和进化成新功能的能力与它们固有的构象丰富的动态性质有关。酶表现出高度的灵活性和多功能性,这表现为它们混杂的侧活性以及它们进化向新功能的耐受性。
酶循环有以下步骤:(1)首先,底物与催化口袋结合,这通常需要和/或诱导环构象h和调节活性位点的进入灵活域的改变,(2)底物(s)被激活,以促进酶−底物(ES)复合物的生产性形成,(3)接下来是形成多个反应中间体和产物的过渡态(s)的稳定(4)最后,一旦酶−产物(EP)复合物形成,产物(s)从口袋中释放出来,这通常伴随着构象变化,引发下一轮催化循环。所有这些步骤对于通过优化整个途径的吞吐量来最大化催化活性都是必要的。底物的结合形成ES也可以调节构象景观,如多酶复合物丙酮酸脱氢酶复合物所示。在图1中,腺苷酸激酶(AdK)催化过程中发生的构象变化。催化循环包括覆盖活性位点的盖子从开放结构到封闭结构的构象变化。计算步骤的计算步骤沿催化行程(步骤2和3)需要使用QM,混合QM/MM,EVB,这些用于分析结合到解离循环过程的构象变化分析成本比较昂贵。目前的计算策略主要只关注上述的一些特征,导致其在实现高水平的酶活性方面的成功率较低。
3.自由能景观的计算重建
酶在溶液中所采用的构象集合可以在自由能景观(FEL)中表示。自由能(G)与kBT单位中的总体分布的负对数成正比;因此,这个分布中的最大值是FEL中的最小值。因此,FEL提供了关于热力学的关键信息和构象转变的动力学。这些分离不同最小值的能量障碍将决定构象交换的时间尺度:快速的构象变化发生在皮秒到微秒的时间尺度上(这是对酶催化至关重要的环运动),而慢速运动将发生在毫秒到秒之间。
酶可以通过x射线、室温、时间分辨x射线、低温电子显微镜、核磁共振和生物物理技术来提供互补的动力学信息。这些存储在蛋白质数据库(PDB)中的相同酶的多重构象在AF2训练中发挥了重要作用,但在AF2应用于评估生物系统的构象异构性方面也发挥了重要作用(如下所述)。计算方法特别适用于重建FEL:MD模拟通过积分牛顿运动定律来采样种群分布。通过定义一组简化的集体自由度(DOFs),可以将在MD运行中获得的高维数据投影出来,用于概率分布计算,从而进行FEL重建(见图2)。通过不同的降维方案,可以手动或自动地选择降维自由度集。对FEL重建的构象变化的准确探索需要广泛的MD模拟,并且根据构象转变的时间尺度,需要应用增强的采样技术。这些技术相关的计算成本很高(从几周到好几个月的模拟),这限制了这些策略在计算设计和排序酶设计中的适用性。

图2 自由能景观(FEL)重建过程的示意图表示。MD模拟的高维数据需要简化并投影到一组关键的集体自由度(DOF)中,以进行概率分布计算来重建FFEL。
4.AF2在捕获构象异构性中的应用

图3 用AlphaFold2(AF2)预测交替状态的策略概述。正如在del Alamo等人所做的那样,多序列比对(MSA)深度可以被改变,一些MSA位置可以被掩盖,如Stein和McHaourab所示,MSA可以聚类。
标准的AF2协议需要酶的一级序列,由进化相关蛋白信息生成的多序列比对(MSA),以及少量命名为模板的同源结构的三维坐标(见图3)。虽然AF2被设计用来预测单个静态结构,但最近的一些论文表明,通过减少AF2算法中使用的输入MSA的深度(除了减少回收的数量),可以生成多种构象的精确模型。del Alamo和他的同事表明,通过改变AF2管道,减少MSA序列(只有16个序列),可以获得转运体和g蛋白偶联受体的多种构象。他们为每种MSA大小每种蛋白受体生成多达50种不同的构象,而标准的AF2协议提供了构象同质和几乎相同的结构。有趣的是,他们观察到AF2训练集中包含的蛋白质的有限的构象采样。在另一项研究中,Stein和McHaourab报道了一种通用的方法,将AF2生成的模型基于将MSA中的特定残基替换为丙氨酸或其他残基。AF2用于生成初始构象,根据初始结构中可能的接触点、先验结构信息或主结构内的不确定性区域,对MSA进行了修改。他们发现,将某些氨基酸柱替换为丙氨酸或其他残基,将网络的注意力转向MSA的其他部分,允许AF2基于其他共同进化的残基找到替代构象。其中一个例子是AdK,它经历了一个盖子和一个锁定活性位点的大规模构象变化,如未结合和抑制剂结合的晶体结构所示(见图1)。通过掩蔽部分残基并将其替换为丙氨酸,得到了AdK的闭链和开链。虽然没有一个AF2开放结构达到晶体结构的开放水平(PDB:4AKE),但生成的AF2模型显示了不同水平的闭合,显示了预测描述系统构象异构性的方法的潜力。
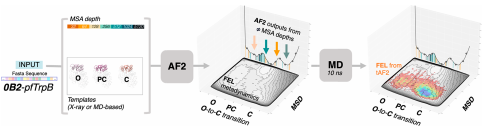
图4 基于模板的AF2(tAF2)方法用于估计构象异质性。不同的多个序列比对(MSA)深度和一组模板从选择x射线结构或分子动力学(MD)快照提供AF2.34多个输出模型由AF2不同MSA深度所示垂直线中心情节然后受到短MD模拟FEL重建。新的FEL从10 ns MD模拟从ca开始产生。在不同MSA下的1000个AF2输出以蓝色到红色的颜色图显示,在通过良好的多步元动力学模拟获得的计算重建的FEL上(灰色)。重建的FELs的x轴和y轴分别表示覆盖活性位点的TrpB的COMM域的开放到封闭(O-to-C)过渡,以及与生成的O-to-C结构路径的均方偏差(MSD)。输入序列是0B2-pfTrpB变体。
受这些工作的影响,作者开发了一种基于模板的AF2方法来评估构象异构性,以及这是如何被几种色氨酸合酶的β亚基突变所改变的(TrpB,见图4)。正如在del Alamo等人的工作中所做的那样,本文测试了减少MSA中提供的序列数量的效果。另外本文评估了当提供不同的显示多个构象状态的模板时,AF2预测是如何改变的。作者通过提供基于X射线或从MD模拟中提取的构象作为模板来测试基于模板的AF2管道。在这些设置下,AF2显示了所分析系统之间构象景观的主要差异。有趣的是,通过从AF2结构集合中运行多个短MD模拟并重建相关的fel,进一步证明了这一点(图4)。从基于模板的AF2预测中生成的FEL与使用良好的多步元动力学模拟生成的计算成本昂贵的FEL相一致。这是令人兴奋的,因为它显示了AF2在快速和准确地评估不同系统的纤维粒体方面的潜力-这可以应用于构象驱动的酶设计方法。AF2通过在不同MSA深度获得的多个输出最近也与重加权自编码变分贝叶斯增强(RAVE)抽样相结合可以获得结果。
5.AF2等深度学习技术在蛋白质和酶设计中的应用
受AF2方法的启发,其他的DL技术最近也被开发出来,用于阐明酶的折叠结构,并提供了一些可能用于蛋白质设计的指标。该领域正在快速发展,特别为蛋白质设计而开发的DL策略的数量也在不断增加。本节的目的是简要概述最具代表性的技术,并特别强调那些与酶设计特别相关的策略。
这些可用的结构预测策略可以根据所使用的输入参数的数量进行分类:那些需要输入查询序列、MSA和用于准确预测的模板集,以及那些仅基于输入序列预测折叠结构的策略。与AF2类似,RoseTTAFold(RF)算法几乎与AF2同时开发,需要一个MSA和一组初始模板来对折叠结构进行准确的预测。RF对−蛋白蛋白的准确性有所提高。OpenFold2也被开发用来复制AF2算法,并使其能够进入结构生物学社区。AlphaLink也被引入,以纳入实验距离约束信息,从而生成了AF2网络架构的修改版本。
序列包含了关于酶的结构和功能的隐式信息,因为每个氨基酸在序列中的位置是由空间排列和它们之间可能建立的相互作用所决定的。其主要优点是序列的比较在计算上很便宜(至少与基于物理的方法相比),并提供了关于每个位置上最常见的残基、保守分数和在进化过程中出现的相关突变对的关键信息。共变突变与功能、3D接触和结合有关。研究还表明,使用以前用于自然语言处理(NLP)的语言模型可以应用于生物学语言的上下文中,从大规模的序列数据集生成“内容感知”的数据表示。这就是ESM-2-它是迄今为止开发的最大的蛋白质序列语言模型。ESMFold可以进行端到端折叠结构预测,在达到与AF2相似的精度的同时速度快一个数量级。OmegaFold(OF)是另一种端到端结构预测算法,它结合了预先训练好的语言模型和几何Transformer模型,用于重建结构。与ESMfold类似,OF只需要输入序列,并且比AF2和RF快10倍。更重要的是,OF在预测孤儿蛋白的折叠结构方面做得更好,即那些没有任何指定功能家族的蛋白质。
除了预测蛋白质折叠结构的不同方法外,不同的NLP和深度学习结构也被开发出来生成新的非自然序列。这些不同的策略针对不同的目标,从生成新的序列来维持一些自然活动到想象新的折叠和复杂的对称组装等。
生成语言模型ProGen和ProGen2在数百万个原始蛋白序列上进行训练以生成表达良好并维持酶功能的从头人工蛋白。ProtGPT2是一种无监督的语言模型,它可以基于自然序列的原理生成新的序列。同样,在荧光素酶样氧化还原酶数据集上训练的变分自编码器也被用于生成维持荧光素酶活性的新序列。ProteinGAN基于生成对抗网络的一种基于自我注意的变体,学习自然蛋白质序列以生成新的功能变体。在BRENDA酶数据库上训练的条件语言模型ZymCTRL最近也被开发出来,它能够在一个用户定义的基于酶分类(EC)的酶类中提供新的人工酶。语言模型也被用于获得一组可能折叠成给定所需结构的序列。例如,最近开发的LM-Design和ProteinDT就是这种情况。Yu和他的同事最近开发了基于对比学习的CLEAN,它能够将EC数分配给给定的序列。
转化约束trRosetta(trRosetta)由贝克实验室于2020年开发,通过随机修改起始序列来找到快速预测的−残基残基间距离图来设计多种蛋白质。trRosetta和基于物理的Rosetta的结合提供更多漏斗的能量景观:trRosetta被用于反对替代状态,高分辨率trRosetta被用于在设计的目标结构上创建一个深度能量最小值。trtrRosetta促进了小β桶蛋白和具有不连续功能位点的蛋白的研究发展。最近,Dauparas和他的同事开发了一种名为ProteinMPNN的方法,这是一种图神经网络,可以挽救以前Rosetta或AF2产生的失败设计。ProteinMPNN最近被用于生成从头荧光素酶。MutComput是一种卷积神经网络(CNN),已成功应用于设计新的聚(对苯二甲酸乙二酸乙酯)解聚水解酶。Anand等人提供了另一个最近的蛋白质序列设计CNN,以生成从头tim-b桶状蛋白主干。全息cnn也被开发用于学习蛋白质微环境的形状,以预测突变对蛋白质复合物的稳定性和结合的影响。
基于使用AF2来预测生成的序列的结构和使用输出的AF2指标来设计新的蛋白质的不同的协议也已被开发出来。AlphaDesign可以从随机序列开始快速预测全新的蛋白质单体。AlphaDesign在设计与特异性靶蛋白结合的蛋白质方面的潜在应用也被显示。AF2还被用于快速和准确的固定主干设计序列,这些序列可以折叠到一个特定的主干。Baker实验室结合ProteinMPNN和AF2,设计具有中心口袋的封闭重复蛋白,并生成对称的蛋白组装。类似地,RF而不是AF2用于设计高亲和力的蛋白结合物或具有预先指定的功能基序的蛋白。RF也有可能预测突变对蛋白质功能的影响。
基于RF的扩散模型(称为RF扩散)由Baker实验室开发。RF扩散可以非常快速和准确地设计受拓扑约束的蛋白质单体、蛋白质结合物、对称低聚物、金属结合蛋白,甚至是含有特定活性位点残基的酶支架。RF扩散的性能在成功率、准确性和速度方面都优于幻觉设计。即使RF扩散没有明确地考虑底物分子,但它可以使用外部电势隐式地建模来引导活性位点口袋的产生。
如引言中所述,催化功能需要底物的结合和产物的释放,而且在许多情况下,酶的活性依赖于辅助因子和金属离子的结合。在这个方向上,基于DL的不同策略也被产生,以将配体、底物和缺失的辅助因子对接到潜在的口袋中。AlphaFill使用序列和结构相似性,将缺失的有机分子和金属离子纳入AF2模型。扩散生成模型扩散模型被设计用来将小分子对接到潜在的蛋白质口袋中。该策略被证明优于以前的传统和DL对接协议。Meller和他的同事还开发了一种基于AF2的策略找到神秘的口袋。DL也被用于寻找蛋白质中过渡金属的潜在定位位点(Metal1D和Metal3D)。基于共同进化的金属网络管道最近也被创建出来,用于预测潜在的金属结合位点。
6.展望
酶催化是一个复杂的多维过程,需要最优的序列和结构来允许底物(s)结合,催化化学步骤和产物(s)释放,并优化发展其功能所需的多重构象。这种高度的复杂性使得酶设计的任务,特别是对非自然反应或高效的底物非常具有挑战性。这篇综述中突出的例子显示了DL技术在允许的序列空间生物约束范围内的情况下生成新的功能变体的潜力。DL策略在任何目标反应和非天然底物的计算酶设计中的应用才刚刚起步。多年来,在计算酶设计中,将所需的活性位点残基合并到蛋白质支架中缺乏精确性一直被认为是整个过程的许多限制之一。然而,这一点似乎可以用Baker实验室最近开发的基于RosettaFold的扩散模型来解决。QM-based模型的酶活性网站到新的非天然支架专门设计的功能主题可能不再是限制因素,而是预测哪些支架可能更适合的优化构象集成有效催化最有可能是必不可少的。考虑到巨大的进步特别是在结构预测和蛋白质设计领域近年来,DL方法结合物理方法将发挥关键作用,未来几年找到最优解决方案的理性和常规设计高效和稳定的酶非天然反应和基质。
-------------------------------------------
欢迎点赞收藏转发!
下次见!















 4083
4083











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









