






我要给你安利一个R语言绘图的超实用干货——集合可视化的神器:UpSetR包!🌟
它能够优雅地处理集合间的交集、并集,让数据的对比和关系一目了然。🎨告别那些让人眼花缭乱的传统图表,用UpSetR包让你的数据图形简洁又美观,还能轻松展示出更多的信息。✨
🚀 特点速览:
1️⃣ 直观展示集合关系
2️⃣ 动态交互,探索数据更深入
3️⃣ 自定义设置,满足你的个性化需求
4️⃣ 一键导出,分享你的发现
🔬 数据分析,不再只是数字游戏,让我们一起用UpSetR包,把数据变成故事,讲述属于你的见解。📚
关于不同集合之间的交集关系,相信大家都常见到。
而最常用于了解交集的图表应该就是文氏图(Venn diagram)【往期文章:文氏图_1; 文氏图_2】,可是一旦集合数超过4个以上,文氏图解读起来就没那么轻松了。因为3个集之间的可能交集只有8种,但如果是10个集,它们之间的可能交集高达1024种(即 2n )![1]。
此时就可以考虑使用UpSet plots[2]。
今天介绍一个发表在《Bioinformatics》上的R包{UpSetR}[3-4],轻松处理3个以上集合的交集关系。
首先,安装R包并且载入:
install.packages("UpSetR")
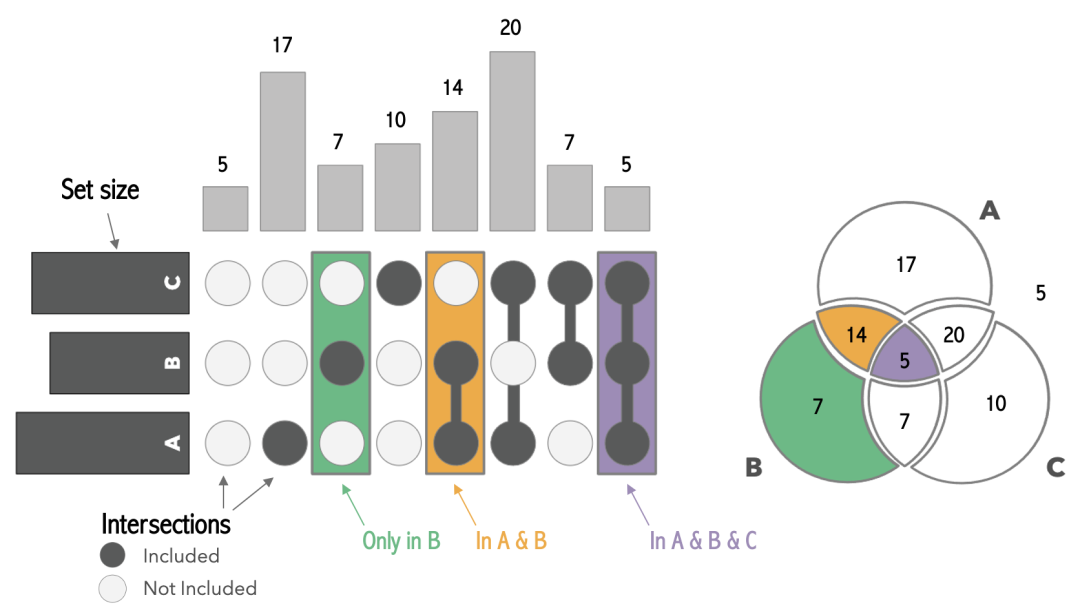
library(UpSetR)UpSet图是一种用于展示不同组之间共有和特有部分的可视化方式,它是韦恩图的另一种替代方式。UpSet图可以用于展示3-7组(目前设置最大的是7组)之间的共有和独有的情况。UpSet图主要包含三个部分:上部分为各个分组独有和共有的数量,下部分为各个分组独有和共有的分类情况,左部分(或者右部分)代表各个分组中包含的没有重复的元素数量。每一列都是一个分组,而每一行则代表一个元素。
在UpSet图中,可以通过柱状图和点的结合来展示各个分组之间的交集情况。柱状图的高度表示某个分组中独有或共有的元素数量,而点则用于表示元素在各个分组中的分布情况。通过点的位置和连接关系,可以直观地看出各个分组之间的交集情况。
在R中,可以使用UpSetR包来绘制UpSet图。UpSet图的优势在于它可以展示多个分组之间的交集情况,并且可以直观地比较各个分组之间的差异和相似之处。此外,UpSet图还可以通过排序和筛选等功能来进一步优化可视化效果。
论文
A highly conserved core bacterial microbiota with nitrogen-fixation capacity inhabits the xylem sap in maize plants
https://www.nature.com/articles/s41467-022-31113-w
本地pdf s41467-022-31113-w.pdf
用到的是UpSetR包:https://github.com/hms-dbmi/UpSetR。 upset
参数:
nsets: 最多展示多少个集合数据。
nintersects: 展示多少交集。
mb.ratio: 点点图和条形图的比例。
order.by: 交集如何排序。这里先根据freq,然后根据degree
decreasing: 变量如何排序。这里表示freq降序,degree升序
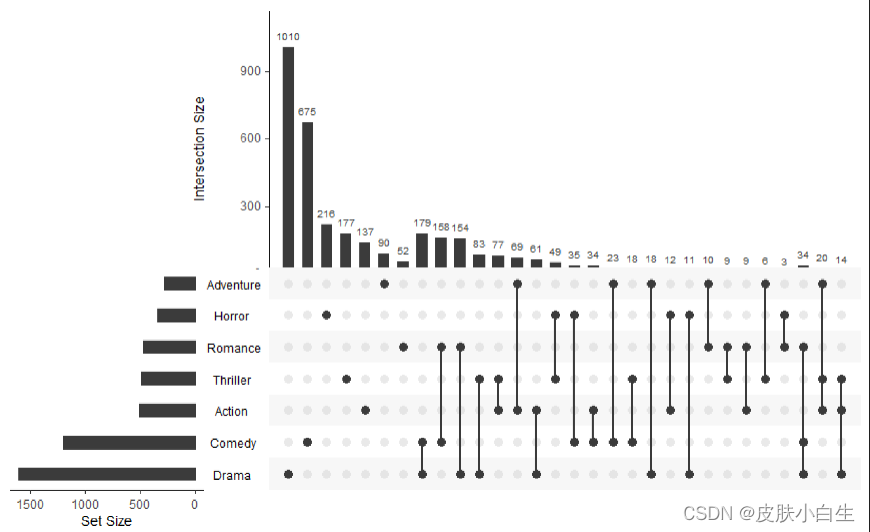
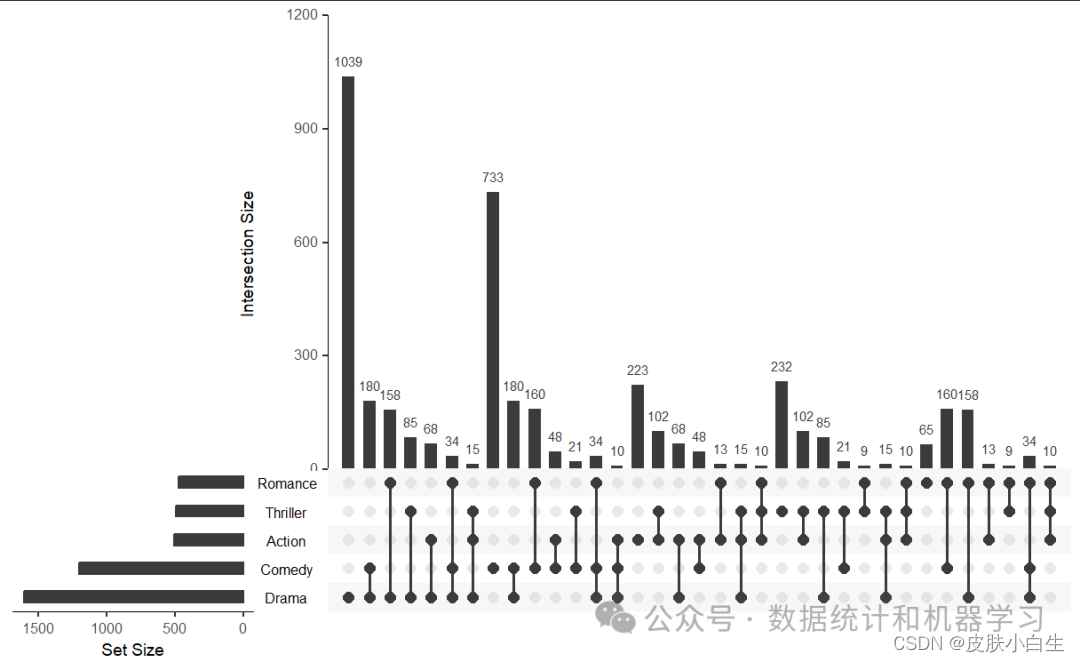
使用的是自带的Demo数据,name是的电影名,第二列是电影的发布时间,后面跟着十几个电影类型,最后两列是average rating以及观看数量,这里需要注意的是upset绘图选取的列只使用二进制列(仅包含 0 和 1)和逻辑列,这里也就是电影类型。
图形解读:黑色表示该位置有数据,灰色的点表示没有。不同点连线表示存在交集。具体数据可以看上面的条形图。不同类型的数据的总量看左边的条形图。
install.packages("UpSetR")
library(UpSetR)
library(ggplot2)
library(plyr)
library(gridExtra)
library(grid)
movies <- read.csv(system.file("extdata",
"movies.csv",
package = "UpSetR"),
header=TRUE, sep=";" )
head(movies)
upset(movies,
nsets = 7,
nintersects = 30,
mb.ratio = c(0.5, 0.5),
order.by = c("freq", "degree"),
decreasing = c(TRUE,FALSE))
ComplexHeatmap包里面有一个很好用的函数:list_to_matrix(),能够直接把列表转换成二进制矩阵,简单演示一下
library(ComplexHeatmap)
lt = list(set1 = c("a", "b", "c"),
set2 = c("b", "c", "d", "e"))
list_to_matrix(lt)图形精细化:
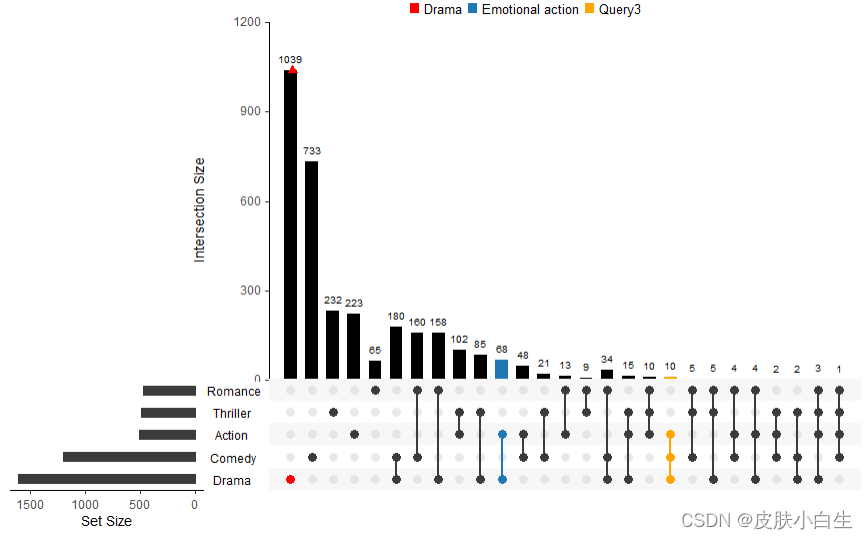
queries参数分为四个部分:query, param, color, active;
query: 指定哪个query,UpSetR有内置,也可以自定义;
param: list, query作用于哪个交集
color:每个query都是一个list,里面可以设置颜色,没设置的话将调用包里默认的调色板;
active:被指定的条形图:TRUE显示颜色,FALSE在条形图顶端显示三角形
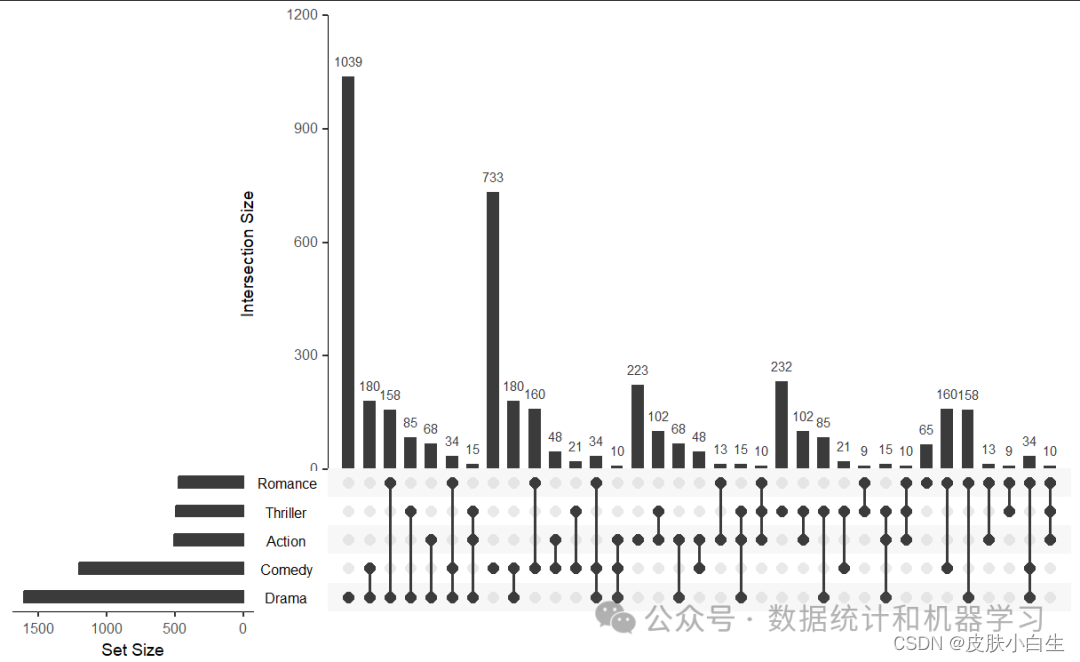
upset(movies, main.bar.color = "black",
queries = list(list(query = intersects, #UpSetR 内置的intersects query
params = list("Drama"), ##指定作用的交集
color = "red", ##设置颜色,未设置会调用默认调色板
active = F, # TRUE:条形图被颜色覆盖,FALSE:条形图顶端显示三角形
query.name = "Drama"), # 添加query图例
list(query = intersects, params = list("Action", "Drama"), active = T,query.name = "Emotional action"),
list(query = intersects, params = list("Drama", "Comedy", "Action"), color = "orange", active = T)),query.legend = "top")
还可以添加一些属性图
upset(movies, main.bar.color = "black",
queries = list(list(query = intersects, params = list("Drama"), color = "red",
active = F, query.name = "Drama"),
list(query = intersects, params = list("Action", "Drama"), active = T,query.name = "Emotional action"),
list(query = intersects, params = list("Drama", "Comedy", "Action"), color = "orange", active = T)),
attribute.plots = list(gridrows = 45, #添加属性图
plots = list(
list(plot = scatter_plot, #散点图
x = "ReleaseDate", y = "AvgRating", #横纵轴的变量
queries = T), #T 则显示出上面queries定义的颜色
list(plot = histogram, x = "ReleaseDate", queries = F)),
ncols = 2), # 添加的图分两列
query.legend = "top") #query图例在最上方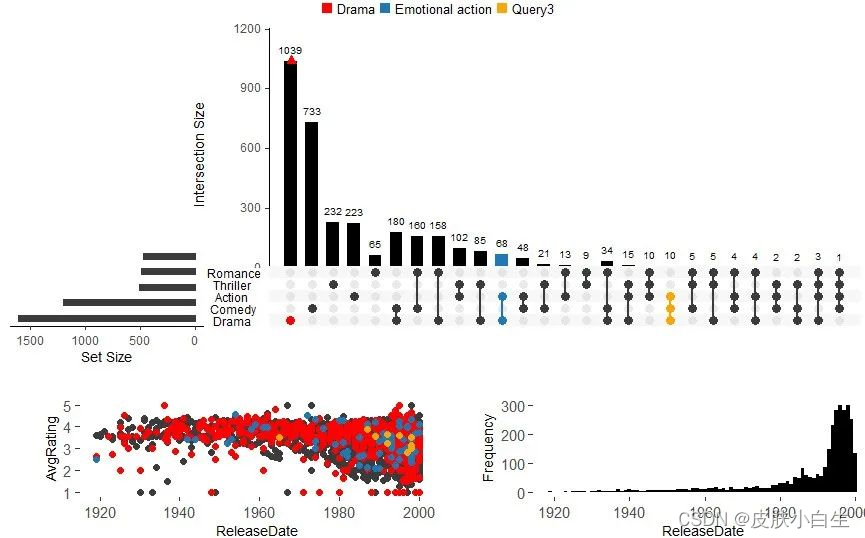
最后来个简单的个性化
参考文章:
https://zhuanlan.zhihu.com/p/370210775 https://www.jianshu.com/p/2347447ea7e4 https://www.jianshu.com/p/d004939b006b https://www.jianshu.com/p/76bfa74bb985 https://www.jianshu.com/p/324aae3d5ea4
数据代码链接
https://github.com/PlantNutrition/Liyu
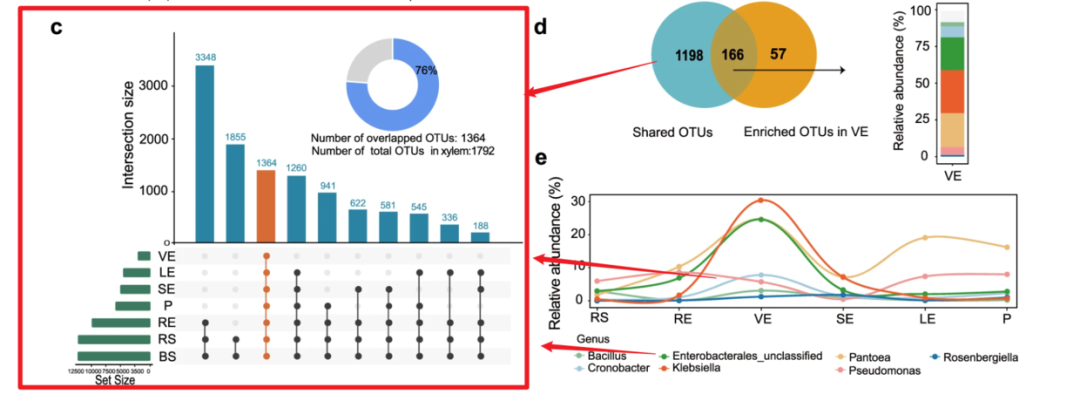
今天的推文我们重复一下论文中的Figure2c
首先是输入数据的格式
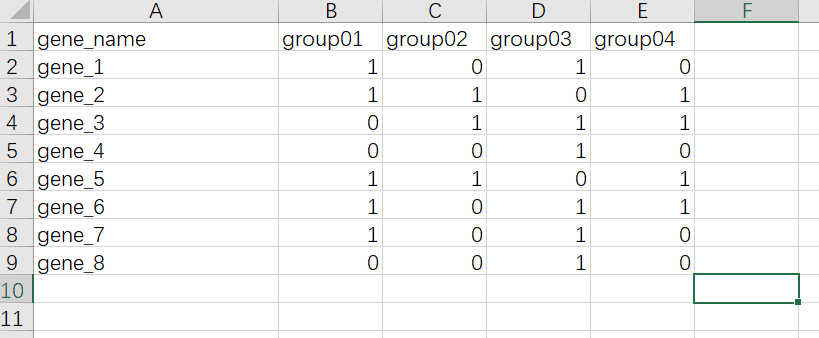
第一列是所有的基因名,读取数据后要将其转换成行名
后面每一列是数据分组,如果这个基因存在于这一组,就标识为1,如果不存在就标识为0
读取示例数据
library(tidyverse)
library(readxl)
dat01<-read_excel("data/20220618/example_upsetR.xlsx") %>%
column_to_rownames("gene_name")
dat01
作图代码
library(UpSetR)
upset(dat01)
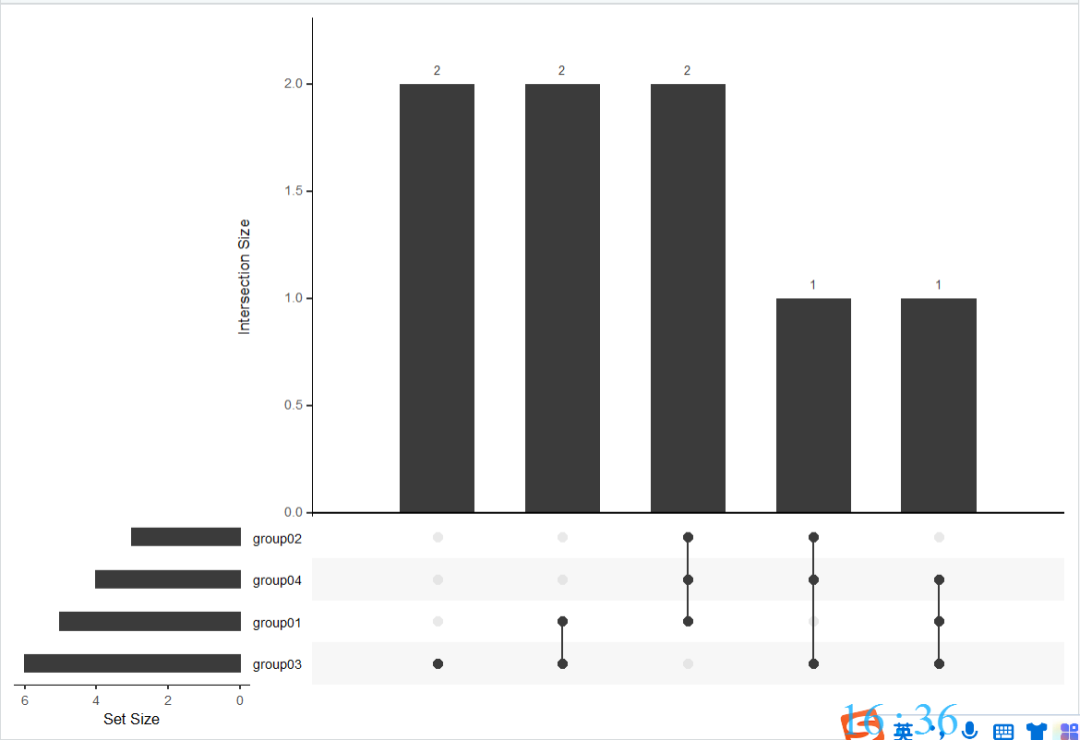
image.png
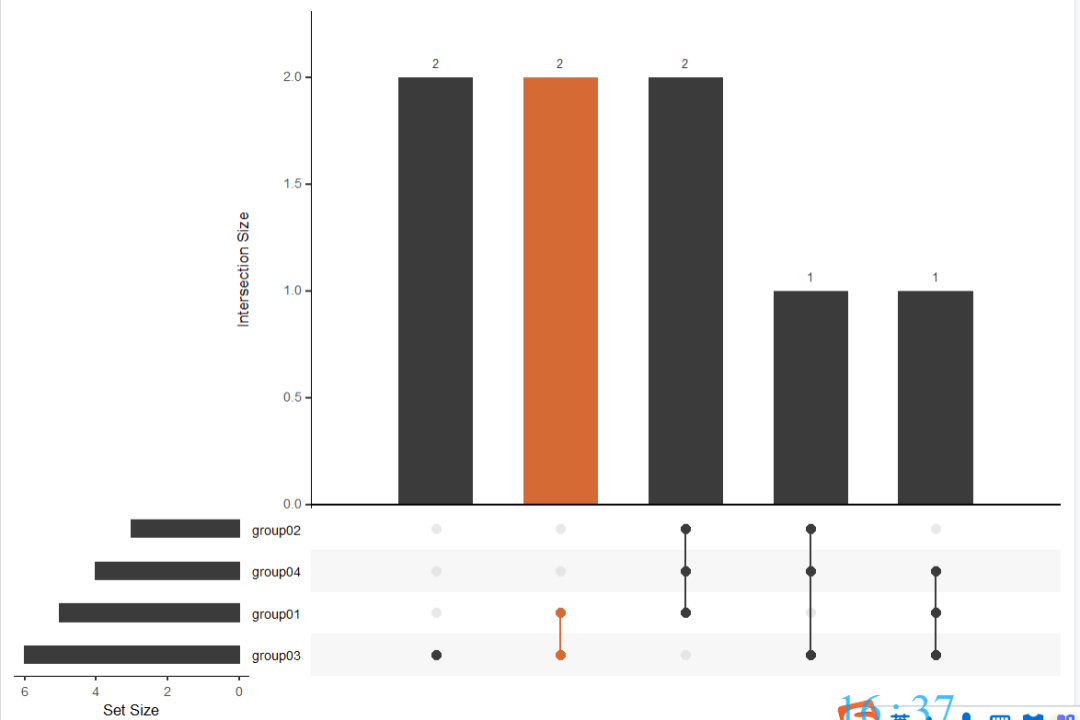
如果要突出强调某一组
queries = list(list(query = intersects,
params = list("group01","group03"),
active = T,
color="#d66a35",
query.name = "ABC"))
upset(dat01,
queries = queries)
这个图主要由三部分组成,让我们通过一个简化的日常生活场景来解释这个图。
1.加载R包和数据
library(UpSetR)## Warning: 程辑包'UpSetR'是用R版本4.1.3 来建造的library(grid)library(plyr)# 构建虚拟数据集listInput <- list(one = c(1, 2, 3, 5, 7, 8, 11, 12, 13),two = c(1, 2, 4, 5, 10),three = c(1, 5, 6, 7, 8, 9, 10, 12, 13))expressionInput <- c(one = 2, two = 1, three = 2,`one&two` = 1,`one&three` = 4,`two&three` = 1, `one&two&three` = 2)# 导入数据movies <- read.csv(system.file("extdata", "movies.csv",package = "UpSetR"),header = T,sep = ";")
2.基本图形
upset(fromList(listInput), order.by = "freq")# or# upset(fromExpression(expressionInput), order.by = "freq")
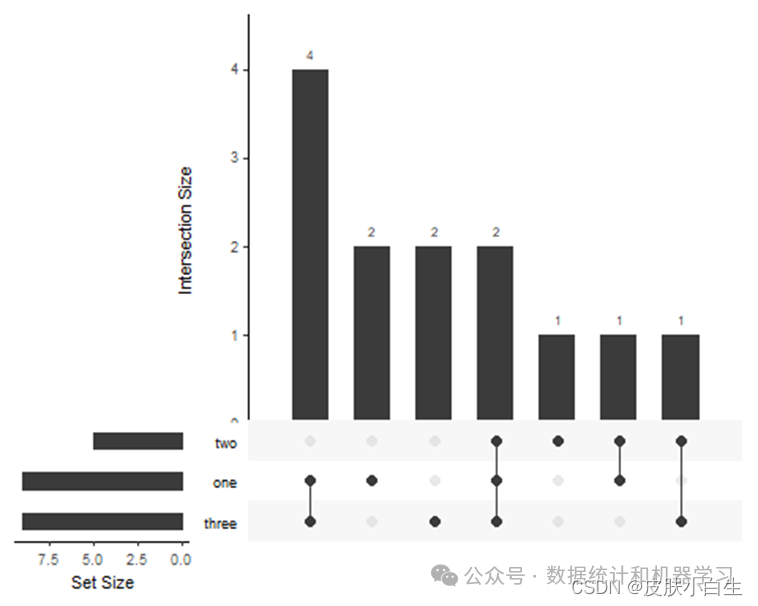
UpSet图由三部分组成:
-
左边的条形图(每个集合中的样本量)
-
上面的条形图(交集的样本量)
-
下面的点和线(显示不同集合之间的交集)
3.图形调整
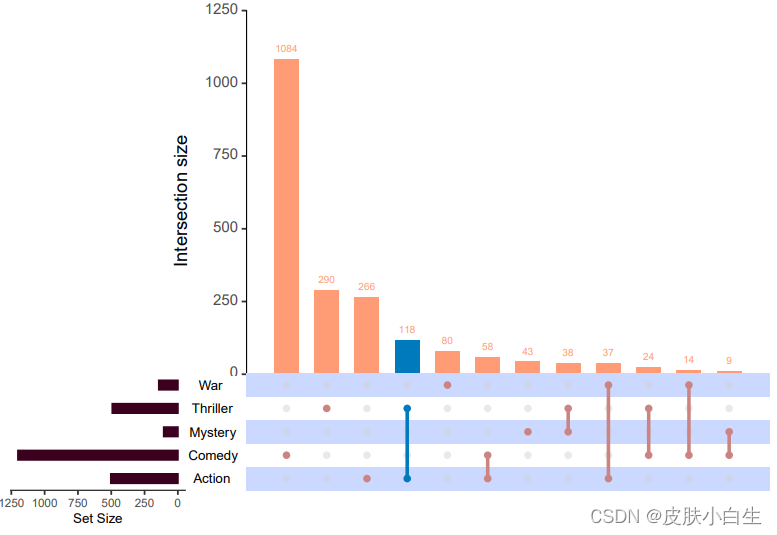
upset(movies,nsets = 6, # 集合数量number.angles = 30, # 数字倾斜角度point.size = 3.5, # 点缩放比例line.size = 2, #线条缩放比例mainbar.y.label = "Genre Intersections",sets.x.label = "Movies Per Genre")
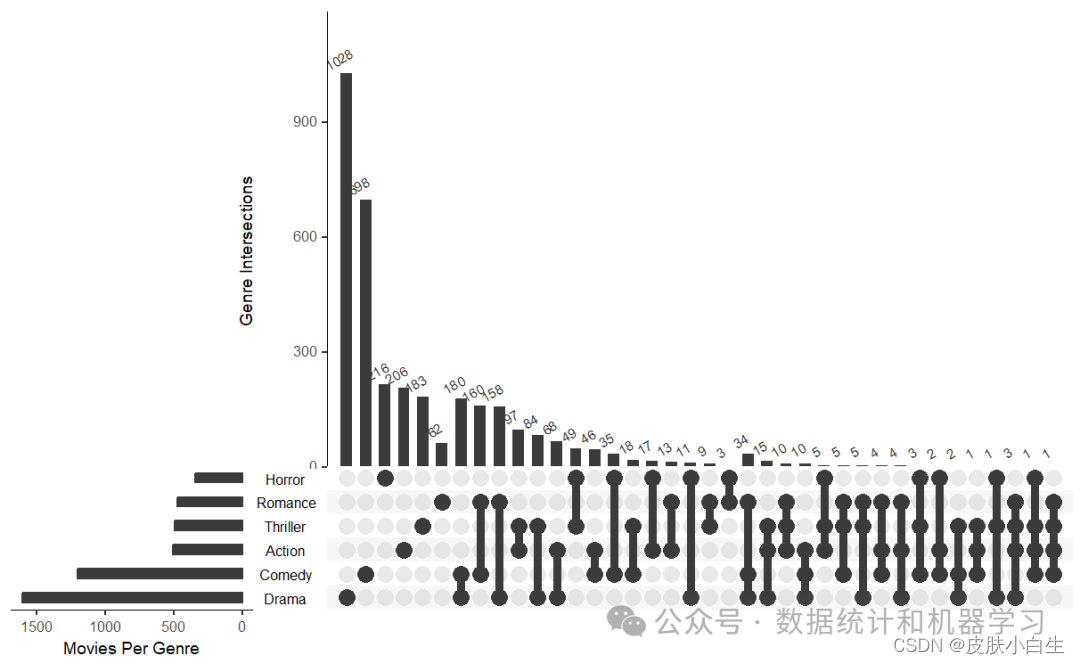
4.选择特定的集合进行比较
upset(movies, sets = c("Action", "Adventure","Drama", "Mystery","Thriller", "Romance","War"), # 指定需要比较的集合mb.ratio = c(0.55, 0.45), # 指定上下图形的比例order.by = "freq")
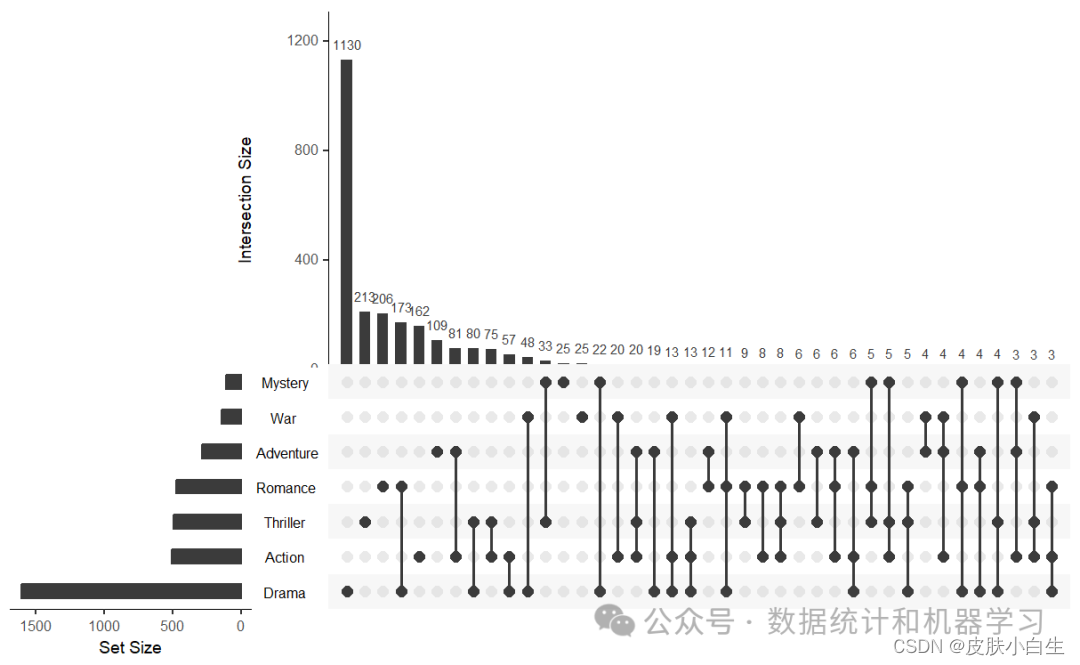
5.设置排序方式
默认的排序方式为“freq”,也可按照“degree”排序。
upset(movies, sets = c("Action", "Adventure","Comedy","Drama", "Mystery","Thriller", "Romance","War", "Western"), mb.ratio = c(0.55, 0.45),order.by = "degree") #设置排序方式
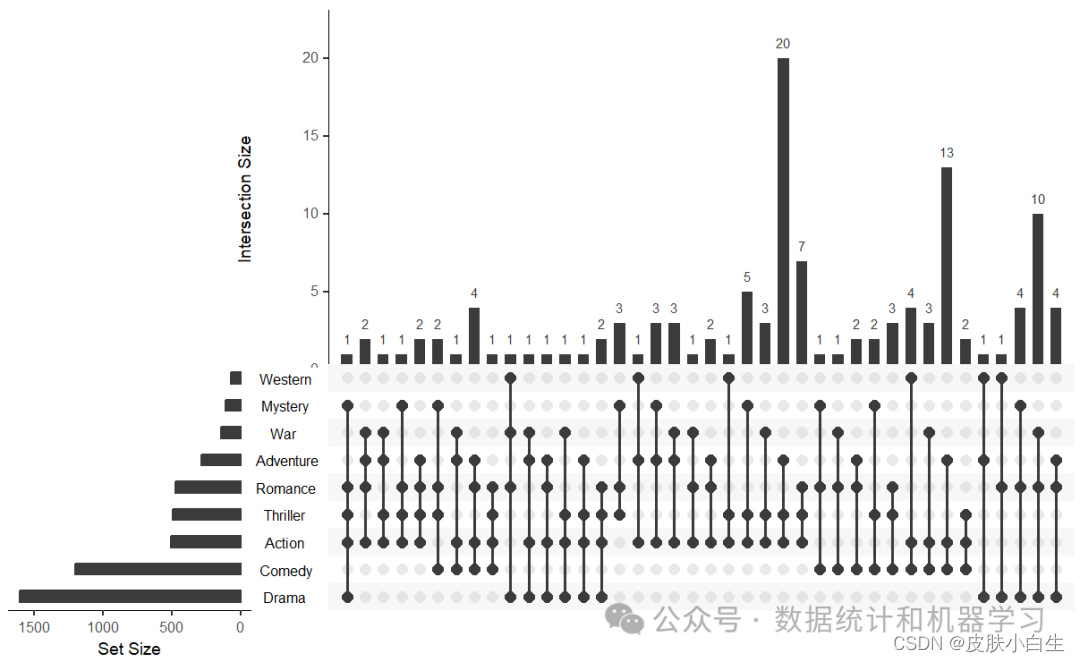
6.分组排序
upset(movies,nintersects = 70,group.by = "sets",cutoff = 7)
7.添加箱型图
upset(movies,boxplot.summary = c("AvgRating","ReleaseDate") # 设置箱型图)
以上只是UpSetR包的基础应用,在此基础上可以修改线条、点、条形图的颜色及大小进行美化;其它的应用请参考官方文档。
参考资料
https://github.com/hms-dbmi/UpSetR?tab=readme-ov-file
otu_RA <- read.delim('example_data/09-venndiagram/otu_RA.txt', header = TRUE, row.names = 1, sep = '\t')
head(otu_RA)
otu_RA[otu_RA > 0] <- 1
head(otu_RA)
他这里把otu表格里有数值的就变成1,只要有数值就说明这个样本中有这个otu
list(list(query=intersects,
params=list("RS","BS"),
active=T,
color="red"),
list(query=intersects,
params=list("RS","BS","RE"),
active=T,
color="blue")) -> queries
upset(otu_RA,
nset = 7,
nintersects = 10,
order.by = c('degree','freq'),
decreasing = c(FALSE, TRUE),
mb.ratio = c(0.5, 0.5),
point.size = 1.8,
line.size = 1,
mainbar.y.label = "Intersection size",
sets.x.label = "Set Size",
main.bar.color = "#2a83a2",
sets.bar.color = "#3b7960",
queries = queries)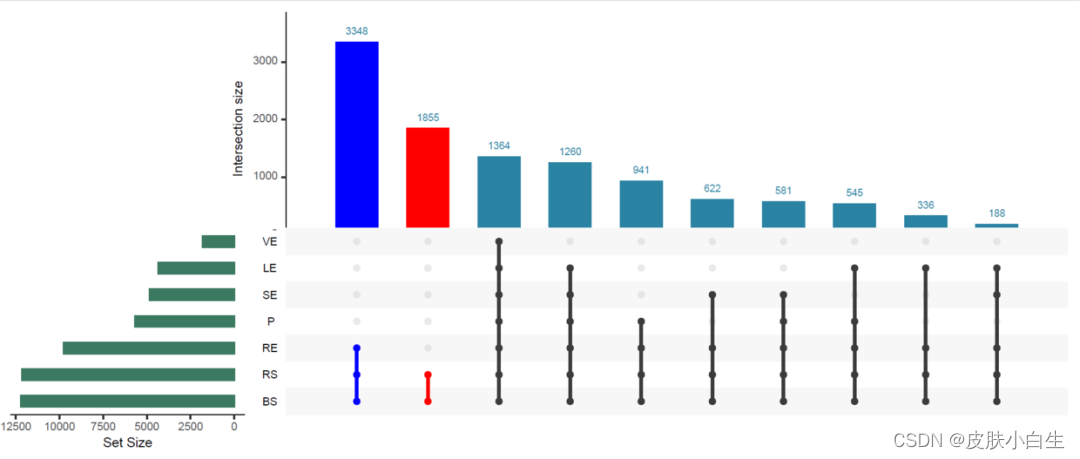
示例数据和代码可以到论文中去下载,或者直接在公众号后台留言20220618获取
明天下午两点半到3点半直播分享R语言ggplot2科研数据可视化入门的一些基础内容,感兴趣的可参考文档链接https://rpubs.com/xiaoming24/916001
示例COVID-19六种临床症状的情况。
如果有该症状则为1,无症状则为0:
set.seed(666)
mydata <- data.frame(
Anosmia = sample(c(0, 1), 666, replace = T, prob = c(0.2, 0.8)),
Cough = sample(c(0, 1), 666, replace = T, prob = c(0.3, 0.7)),
Fatigue = sample(c(0, 1), 666, replace = T, prob = c(0.1, 0.9)),
Diarrhea = sample(c(0, 1), 666, replace = T),
Breath = sample(c(0, 1), 666, replace = T, prob = c(0.7, 0.3)),
Fever = sample(c(0, 1), 666, replace = T, prob = c(0.45, 0.55))
)
str(mydata) # 查看数据格式
下一步,使用R包{UpSetR}作图:
【代码显示不全时,可左右滑动】
upset(mydata, # 数据
nsets = 6, # 六个集
number.angles = 10, # 柱状图上数字旋转的角度
order.by = "freq", # 按频次排序
mainbar.y.label = "Intersection sizes", # 修改上方柱状图y轴名
sets.x.label = "Numbers of symptoms", # 左下sets的x轴名
point.size = 3, # 点的大小
line.size = 0.5, # 线的粗细
shade.color = "grey", # 点的背景阴影颜色
matrix.color = "grey60", # 点的颜色
mb.ratio = c(0.55, 0.45), # 上、下两部分图的占比
main.bar.color = "cadetblue4", # 上方图柱的颜色
sets.bar.color = "grey60", # 左下图柱的颜色
text.scale = 1.1) # 图上文字的大小,可分开调整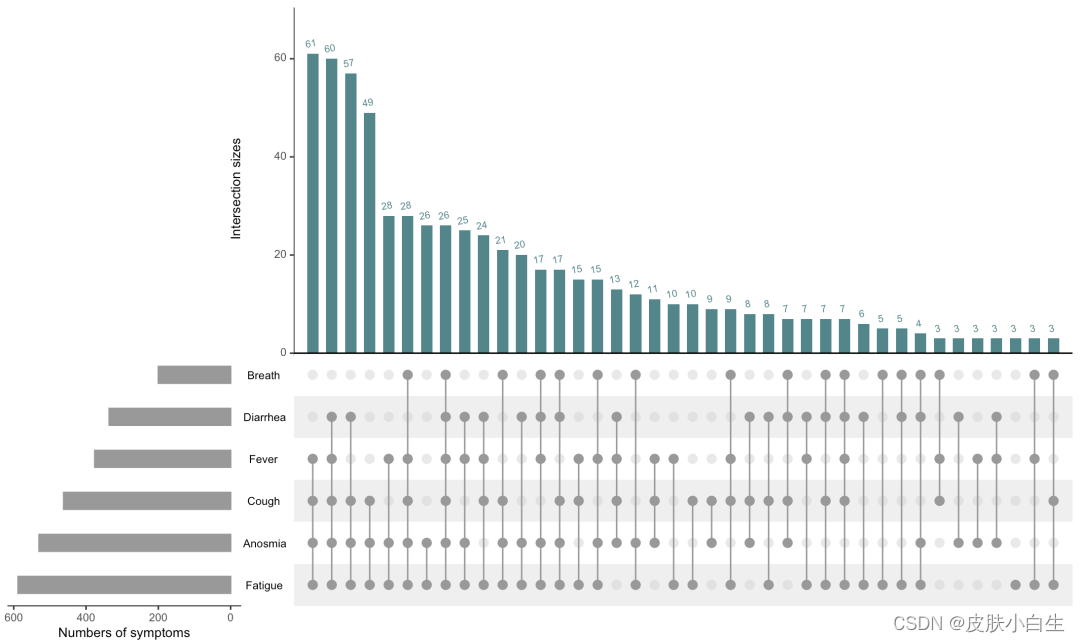
如上图,上方柱状图代表每个交集中样本的个数,按频次从大到小排序;左下为六个集(sets)的数目;右下为相互交集的关系图,由点和线组成。上述示例代码中只调整了R包中的部分函数,感兴趣可以自行深入摸索
最后,小编还制作了一个“UpSet图解”,解释不同部分的含义[2],并且与文氏图(Venn diagram)进行对照:
交集基因
链接: https://pan.baidu.com/s/1XIn-sX07c-LQpxLqgwGMUQ?pwd=kzd4
提取码: kzd4
一、安装和加载"UpSetR"包
下载"UpSetR"包
install.packages("UpSetR")加载UpSetR包
library(UpSetR)二、设置工作目录
设置工作目录
setwd("D:\\demo\\UpSerR")三、数据整理
输出交集基因文件
outFile="intersectGenes.txt"输出图片
outPic="upset.pdf"获取目录下所有文件
files=dir()提取.txt结尾的文件
files=grep("txt$",files,value=T)geneList=list()
读取所有.txt结尾文件中的基因信息,保存到geneList
length(files)读取文件夹中所有gene长度
for(i in 1:length(files)){inputFile=files[i]if(inputFile==outFile){next}rt=read.table(inputFile,header=F) 读取输入文件geneNames=as.vector(rt[,1]) 提取基因名称geneNames=gsub("^ | $","",geneNames) 去掉基因首尾的空格uniqGene=unique(geneNames) 基因取unique,唯一基因列表header=unlist(strsplit(inputFile,"\\.|\\-"))geneList[[header[1]]]=uniqGeneuniqLength=length(uniqGene)print(paste(header[1],uniqLength,sep=" "))}
四、绘图
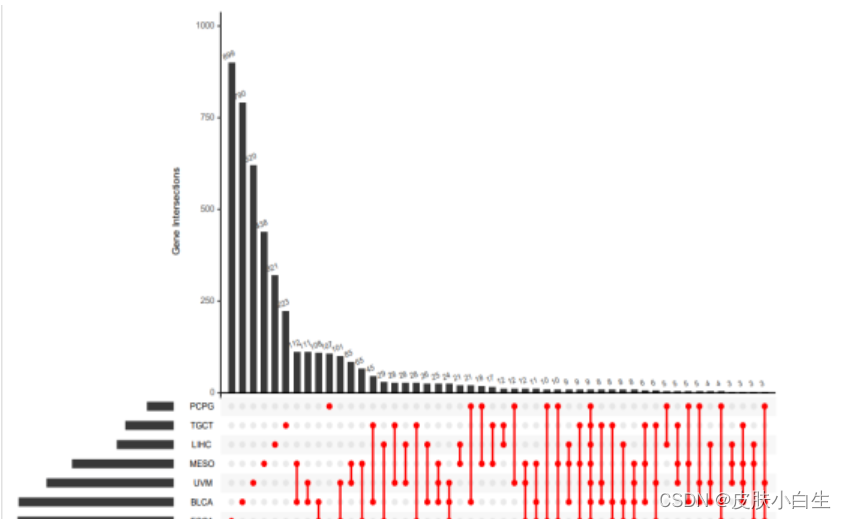
绘制UpSet
upsetData=fromList(geneList)upset(upsetData,nsets = length(geneList), 展示多少个数据nintersects = 50, 展示基因集数目order.by = "freq", 按照数目排序show.numbers = "yes", 柱状图上方是否显示数值number.angles = 20, 字体角度point.size = 2, 点的大小matrix.color="red", 交集点颜色line.size = 0.8, 线条粗细mainbar.y.label = "Gene Intersections",sets.x.label = "Set Size")
五、保存图片、数据
保存图片
pdf(file=outPic,onefile = FALSE,width=9,height=6)dev.off()
保存交集文件
intersectGenes=Reduce(intersect,geneList)write.table(file=outFile,intersectGenes,sep="\t",quote=F,col.names=F,row.



















 515
515

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











