







QIIME2学习
QIIME2分析之单端数据的导入与Deblur
文章目录
前言
当fastq数据质量信息不完整/单一时,无法使用DADA2降噪——DADA2的算法不允许质量是一个值的,DADA2去重复直接是基于自己的数据本身,所以对质量要求严格
而这种数据可以用 Deblur 运行—— Deblur 算法不一样,Deblur 去重复的时候会参考数据库,但对数据库的完整度要求就比较高了,不然可能舍弃掉比较多的序列
为了使用质量信息有问题的fastq数据(前提是数据已经质控过,只是在NCBI上下载的数据质量信息有问题),我们使用 Delbur 替代 DADA2 降噪
一、导入数据
qiime tools import \
--type 'SampleData[SequencesWithQuality]' \
--input-path config.txt \
--output-path mjsample.qza \
--input-format SingleEndFastqManifestPhred33V2
查看文件的原始数据的序列数等信息
qiime demux summarize \
--i-data mjsample.qza \
--o-visualization mjsample.qzv
导出qzv文件查看
qiime tools export --input-path mjsample.qzv --output-path mjsample_statistic
二、Deblur
1.按测序碱基质量过滤序列
time qiime quality-filter q-score \
--i-demux mjsample.qza \
--o-filtered-sequences demux-filtered.qza \
--o-filter-stats demux-filter-stats.qza
输出结果文件:
demux-filtered.qza: 序列质量过滤后结果;
demux-filter-stats.qza: 序列质量过滤后结果统计。
48条序列用时5分多
2.去噪16S过程
deblur去噪16S过程,输入文件为质控后的序列,设置截取长度参数,生成结果文件有代表序列、特征表、样本统计。
time qiime deblur denoise-16S \
--i-demultiplexed-seqs demux-filtered.qza \
--p-trim-length 300 \
--o-representative-sequences rep-seqs-deblur.qza \
--o-table deblur-table.qza \
--p-sample-stats \
--o-stats deblur-stats.qza
输出结果文件:
deblur-table.qza 特征序列丰度表
rep-seqs-deblur.qza 特征序列文件
deblur-stats.qza 质控、去噪、去嵌合体后剩下的序列文件#查看每个样品剩余有效序列数(丰度表)—统计的文件
此处时间长,48条序列用时37分钟
3.输出文件可视化
质控、去噪、去嵌合体的统计结果#打开deblur-stats里的index.html查看
time qiime metadata tabulate \
--m-input-file demux-filter-stats.qza \
--o-visualization demux-filter-stats.qzv
time qiime deblur visualize-stats \
--i-deblur-stats deblur-stats.qza \
--o-visualization deblur-stats.qzv
qiime tools export --input-path deblur-stats.qzv --output-path deblur-stats
qiime tools export --input-path demux-filter-stats.qzv --output-path demux-filter-stats
质控、去噪、去嵌合体的统计结果#打开deblur-stats里的index.html查看
其他表格结果查看
质控、去噪、去嵌合体的统计结果#打开deblur-stats里的index.html查看
time qiime feature-table tabulate-seqs \
--i-data rep-seqs-deblur.qza \
--o-visualization rep-seqs-deblur.qzv
time qiime feature-table summarize \
--i-table deblur-table.qza \
--o-visualization deblur-table.qzv \
--m-sample-metadata-file sample.tsv
time qiime tools export --input-path deblur-table.qzv --output-path deblur-table_stat
导出特征序列文件
qiime tools export --input-path rep-seqs-deblur.qza --output-path rep-seqs-deblur
导出txt格式的丰度表
qiime tools export --input-path deblur-table.qza --output-path deblur-table
biom convert -i deblur-table/feature-table.biom -o asv_deblur-table.txt --table-type "OTU table" --to-tsv
4.结果解读
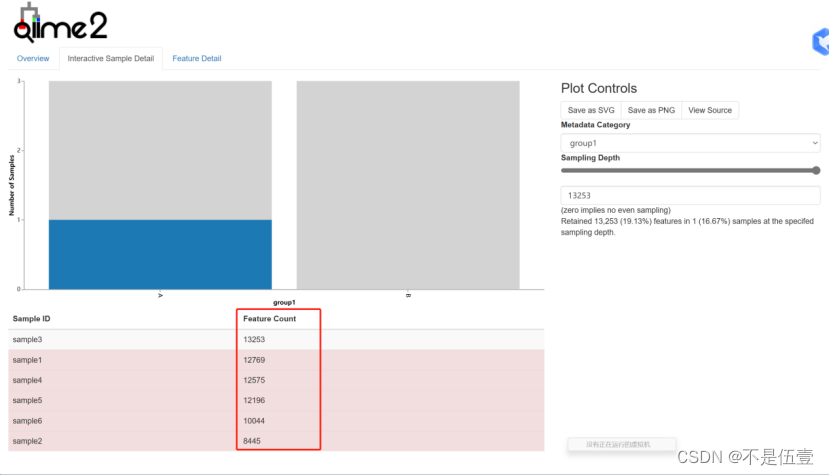 deblur-table_stat中的sample-frequency-detail.html
deblur-table_stat中的sample-frequency-detail.html
框住的一列代表测序深度
多样性时 一般选取最小的;最小值格外小,则去除最小值选最小的
补充说明
如果使用deblur-16S,deblur执行初始的正向过滤步骤,其中它丢弃与85% GreenGenes 数据库中OTU的序列小于60%相似性的任何序列。如果不想执行此步骤,请使用deblur-other方法。
deblur目前只能对单端序列进行去噪。如果提供末合并的双端序列为输入,将对反向序列不作任何操作。请注意,deblur接受合并的序列,并将它们视为单端序列,因此如果使用deblur进行去噪,需要先合并读取。
qiime deblur COMMANDS
Usage: qiime deblur [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin wraps the Deblur software for performing
sequence quality control.
Plugin website: https://github.com/biocore/deblur
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Commands:
denoise-16S:
Deblur sequences using a 16S positive filter.
denoise-other :
Deblur sequences using a user-specified positive filter.
visualize-stats :














 861
861











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









