






SUPPA2 分析可变剪切
1、下载基因组转录本文件
以人类基因组转录本文件下载为例:
打开网站ensemble:http://asia.ensembl.org/index.html --> 点击 human
点击download dna fasta
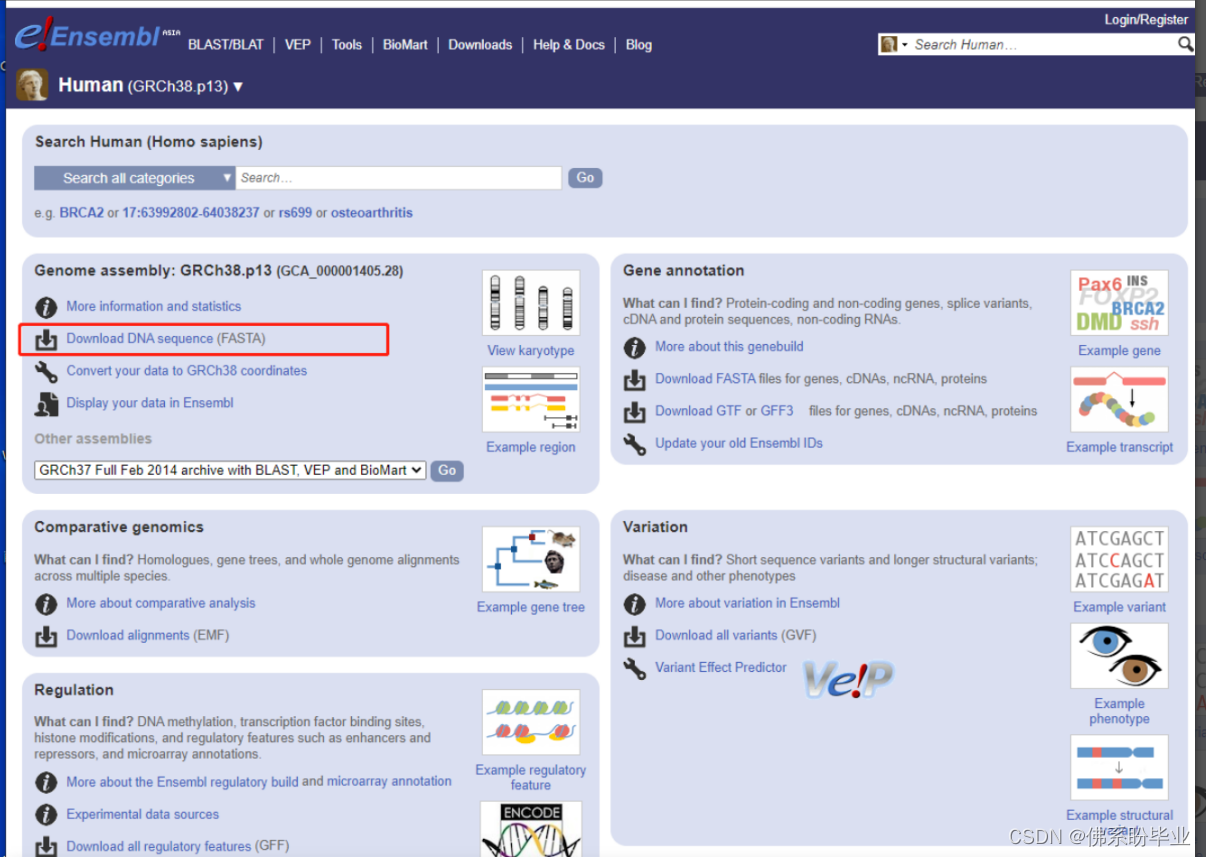
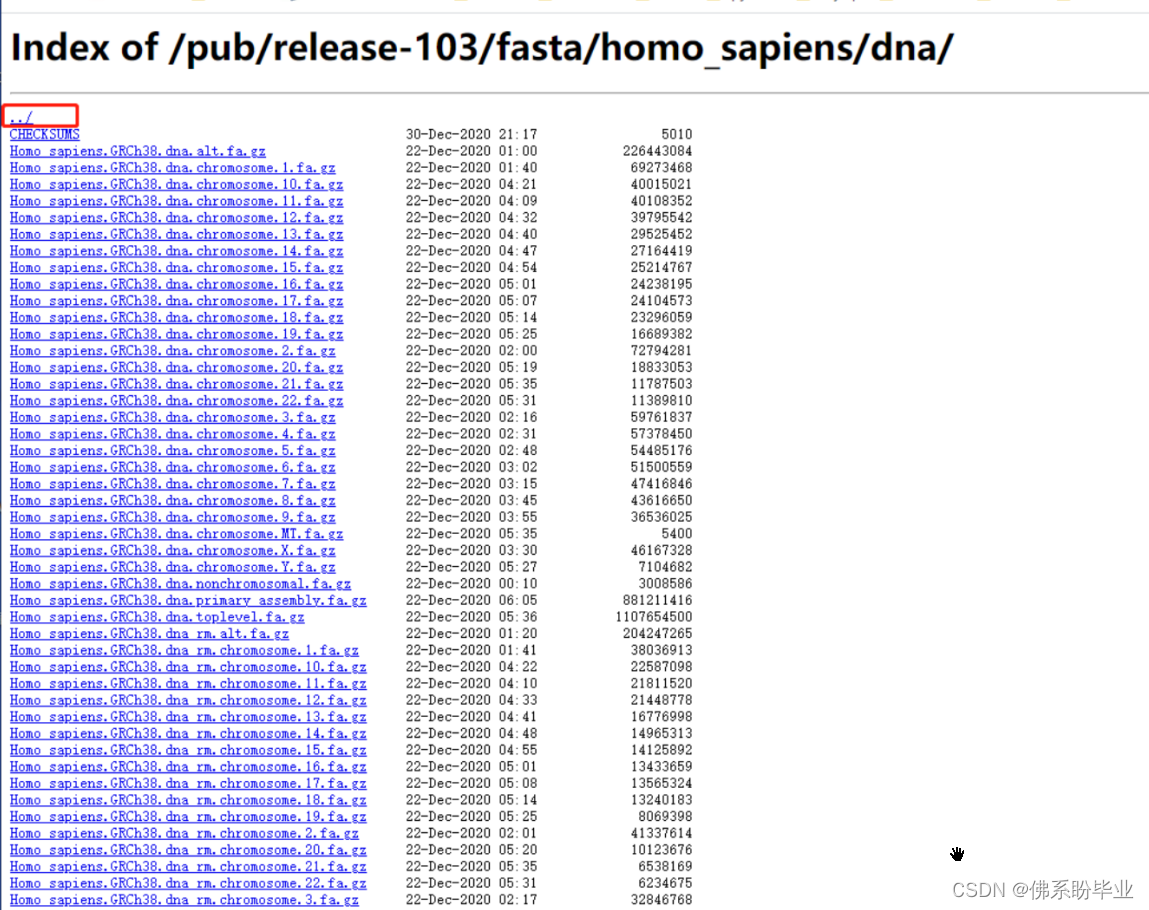
点击划红线的部分,并下载到本地,之后上传到服务器中。
也可以在https://www.gencodegenes.org/mouse/ 网站中下载gtf文件和转录本fa文件
2、解压基因组转录本文件
gunzip Homo_sapiens.GRCh38.cds.all.fa.gz
3、转变为format文件
perl -lane 'if(/^>/){$id=(split/\./,$_)[0];print $id}else{print}' Homo_sapiens.GRCh38.cds.all.fa >Homo_sapiens.GRCh38.cds.all.format.fa
4、创建索引
#创建存放索引的文件夹
mkdir Homo_sapiens.GRCh38.cds.index
#安装定量软件 salmon
conda install salmon
#创建索引
salmon index -t Homo_sapiens.GRCh38.cds.all.format.fa -i Homo_sapiens.GRCh38.cds.index
5、使用salmon进行定量
#对单末端数据进行批量定量 (-r)
ls *gz|cut -d"_" -f 1|sort -u |while read id;do(salmon quant -i /data/gs/data/data1/rawdata3/single/filter-adapters/single-porechop/nanofilt/salmon/Homo_sapiens.GRCh38.cds.index -l ISF --gcBias -r ./${id}_trimmed.fq.gz -o quants/${id}_output);done
#对双末端数据进行批量定量
ls *gz|cut -d"_" -f 1|sort -u |while read id;do(salmon quant -i /data/gs/data/data1/rawdata3/single/filter-adapters/single-porechop/nanofilt/salmon/Homo_sapiens.GRCh38.cds.index -l ISF --gcBias -1 ${id}_1_val_1.fq.gz -2 ${id}_2_val_2.fq.gz -p 4 -o /data/gs/data/suppa/salmon_output/${id});done
#这里定量的fq文件,是通过RNA-SEQ,过滤之后,得到的数据
#salmon也可以直接对sam/bam文件定量,参数为 -a
#salmon也可以直接使用fa文件进行定量,参数为 -t
#salmon使用索引进行定量,参与为 -i
6、提取salmon结果中的TPM值
multipleFieldSelection.py -i SRR*/quant.sf -k 1 -f 4 -o iso_tpm.txt
对tpm文件格式化:
perl -alne '{/(\|.*\|)\t/; ;s/$1//g;s/\|//g;print}' iso_tpm.txt > iso_tpm_formatted.txt
7、下载human的参考基因组注释gtf文件并处理为format格
式:(并解压)
http://ftp.ensembl.org/pub/release-100/gtf/homo_sapiens/
解压:
gunzip Homo_sapiens.GRCh38.100.gtf.gz
8、使用generateEvents命令根据基因组的gtf注释文件生成
所有的可变剪切事件,格式保存为ioe格式(这一步为检测
gtf文件中的可变剪切事件有哪些)
#安装SUPPA
conda install -c bioconda suppa
#计算gtf文件中的as事件
suppa.py generateEvents -i Homo_sapiens.GRCh38.100.gtf -o 107190A.events -e SE SS MX RI FL -f ioe
将不同的可变剪切事件合并成一个结果
vim ioe.awk
# 在vim脚本中写入(按i,即可将vim转变为可编辑状态)
awk 'FNR==1 && NR!=1 { while (/^<header>/) getline; } 1 {print} ' *.ioe > ensembl_mm10.events.ioe(输出文件名,可以修改)
:wq 保存退出 vim脚本
chmod 755 ioe.awk
./ioe.awk
9、计算psi值,根据tpm和gtf的ioe文件
需要注意的是使用的转录本ID和gtf的转
录本ID应该是一致,数目不一样可能会有错误提示:
suppa.py psiPerEvent -i Homo_sapiens.GRCh38.100.events.ioe -e iso_tpm.txt -o SRR_events
10、差异可变剪切基因寻找
#1-56 这个数字为对照组的样本数量
#1-47 这个数字为实验组的样本数量
#这个数字根据自己的样本数据量设定
cut -f 1-56 SRR_events.psi > ./control.psi
cut -f 1-47 SRR1_events.psi > ./treat.psi
cut -f 1-56 iso_tpm_formatted.txt > ./control.tpm
cut -f 1-47 SRR1_tpm_formatted.txt > ./treat.tpm
suppa.py diffSplice -m empirical -gc -i /data/gs/data/data1/reference/gtf/Annotation/suppa/ensembl_mm10.events.ioe --save_tpm_events -p treat.psi control.psi -e treat.tpm control.tpm -o srr2_diffSplice
















 368
368











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









