






我今天在用samtools将比对好的sam文件转换为bam文件时,一直出现多条重复的错误,如下图
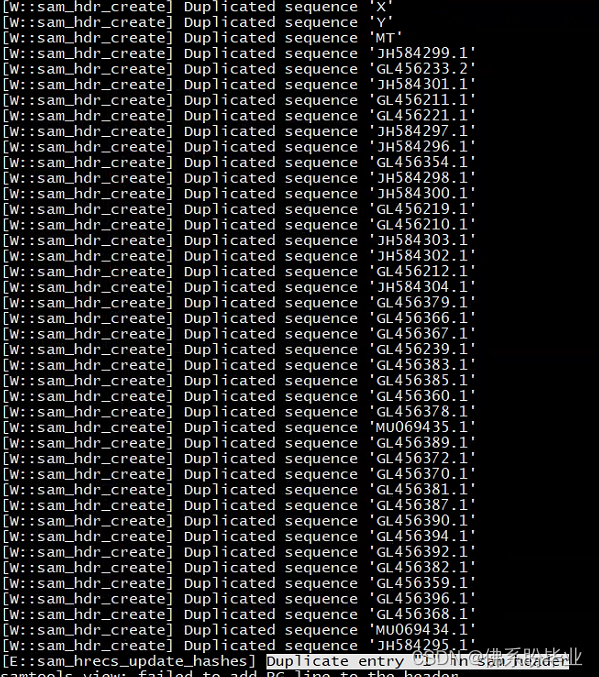
我尝试了很多方法,找到了bamutil这个软件,可以解决这个问题,
但如果只有一条重复,那可以用下面这个方法
#打印重复的内容 && /1/ ,1需要换成你重复的内容值
awk '/^@/ && /1/ { print } /^[^@]/ { exit }' aln-pe.sam
#删除重复的内容 { if (/:1/) 这里面的/:1/,:1需要换成你重复的内容值,冒号要保留,还有:1前后的“/”
awk 'BEGIN { i = 0; } /^@/ { if (/:1/) { if (i++ < 1) { print; } } else { print } } /^[^@]/ { print }' SRR.sam > SRR_fixed.sam
#删除之后,在用samtools转换就可以了
多条重复,可以使用bamutil这个软件
#安装bamutil
conda install -c bioconda bamutil
#sam转为bam
bam convert --in SRR.sam --out SRR_2.bam
#samtools 排序
samtools sort SRR_2.bam -o SRR_2_sorted.bam
#bam转换为无序bam (dedup 输入的文件必须是sorted的)
bam dedup --in SRR_2_sorted.bam --out ./SRR_3.bam
















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









