










1.前言
1.1使用背景
对于主流的单因素cox筛选预后相关基因,以及目前最常见的最小绝对收缩和选择算法——Lasso(Least absolute shrinkage and selection operator)该方法是一种压缩估计。它通过构造一个惩罚函数得到一个较为精炼的模型,使得它压缩一些系数,同时设定一些系数为零。因此保留了子集收缩的优点,是一种处理具有复共线性数据的有偏估计。
Lasso算法最大的特点在于能识别高维变量中的过拟合(Overfitting)现象,通过一系列参数控制模型的复杂度,从而给出一个看似更好、更简易的模型。
1.2算法原理
今天介绍一下随机生存森林模型,看看根据随机生存森林在面对高维基因变量时,如何进行关键基因的筛选,以及森林生存森林建模
随机生存森林(Random survival forest,RSF)的原理为采用自助法(bootstrap)随机选取N个自助样本,对每个自助样本建立二元递归生存树,且每个自助样本保留37%的原始数据作为袋外数据(Out of bag data,OOB)用于模型的测试。最后对每棵树的预测结果进行综合。


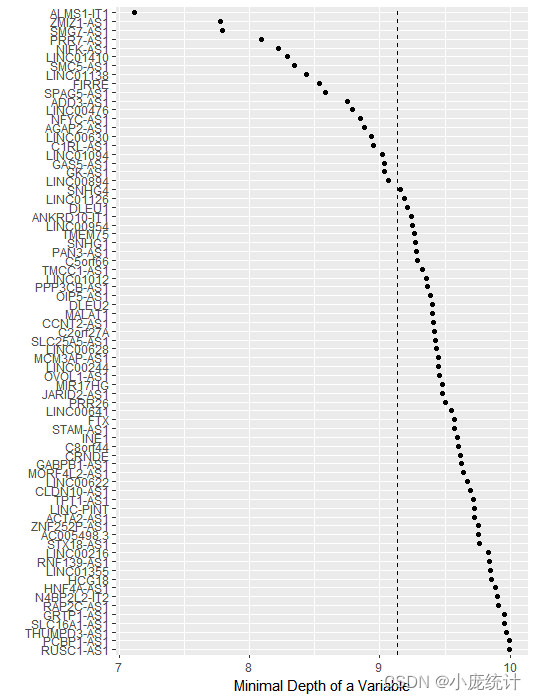
RSF模型的变量筛选通常有两种方法,分别为VIMP法(Variable importance,VIMP)和最小深度法。RSF原理:在纳入一个新变量和不纳入该变量模型错误率的差值。VIMP为正值则说明该变量能增加模型的准确度,如果为负值则说明改变量会降低模型的准确度。最小深度法根据变量是否能区分更多的数据,越靠近根节点的变量越能区分更多变量。因此,最小深度法定义变量重要性根据变量所处节点与根节点之间的距离。换言之,值越小变量越重要。
2.方法
2.1.数据准备
此处的数据为一个转录组的生存数据,其特点是有生存时间,以及生存结局,其次拥有高维变量。
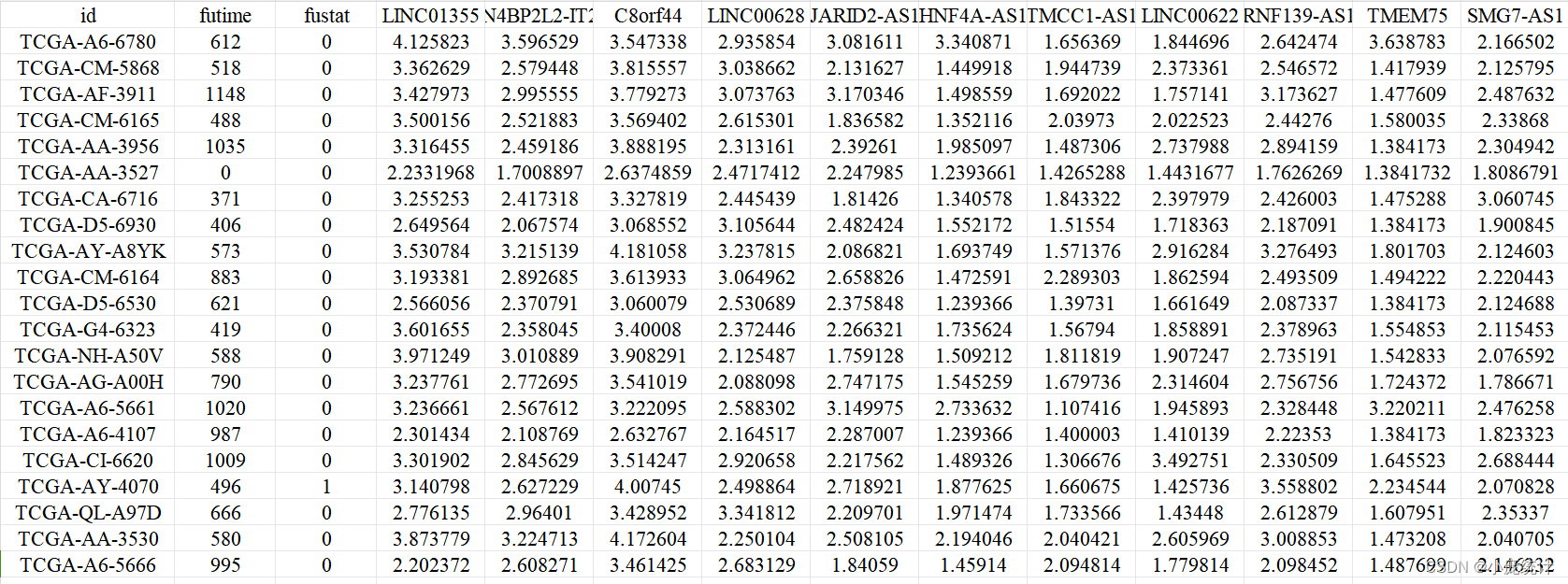
2.2.构建随机生存森林模型RSF
library(randomForestSRC)
rt=read.table(file = "tcga.expTime.txt", header=T, sep="\t", check.names=F)
rownames(rt)=rt[,1]
rt=rt[,-1]
#建立随机生存森林模型
rfsrc_pbcmy <- rfsrc(Surv(futime, fustat) ~ .,
data = rt,
nsplit = 10,
na.action = "na.impute",
tree.err = TRUE,
splitrule='logrank',
proximity = T,
forest = T,
#ntime = 60,#时间节点
#mtry = 10,#变量数
ntree=1000,#树的数量
importance = TRUE#变量重要性VIMP
#block.size=100,#第几颗树的错误率
)2.33.ggRandomForests变量重要性
library(ggRandomForests)
#首先查看VIMP法,排名前15的变量重要性
gg_dta <- gg_vimp(rfsrc_pbcmy, nvar=15)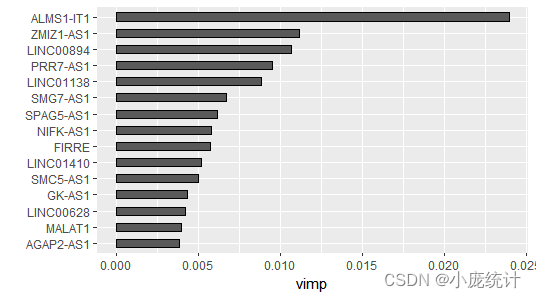
#最小深度法查看变量重要性gg_dta <- gg_minimal_depth(rfsrc_pbcmy)plot(gg_dta)
#两种方法的结合 VIMP+min_depth
gg_dta <- gg_minimal_vimp(rfsrc_pbcmy)
plot(gg_dta) 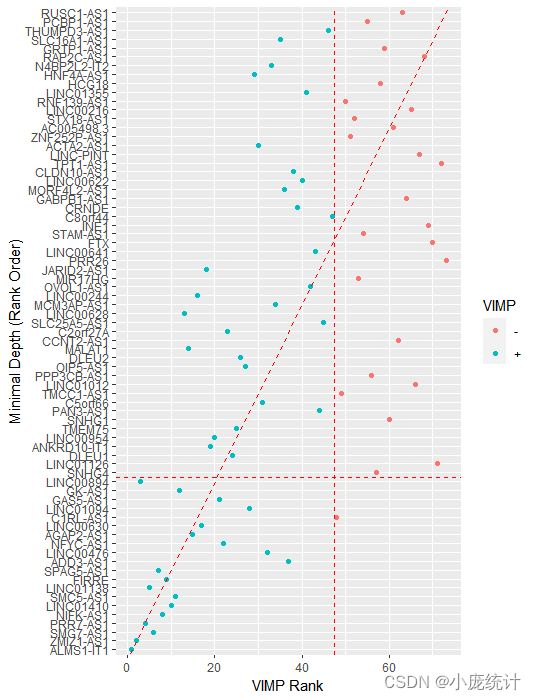
这里我们要根据depth threshold 这个值,来确定筛选出的变量,当小于阈值时就得到了,在VIMP法和最小深度法结合的情况下筛选出的变量。
2.4.筛选变量,构建随机生存森林
filterExp_time是提取重要基因后的文件
rt=read.table(file = "filterExp_time.txt", header=T, sep="\t", row.names=1,check.names=F)
#模型构建
##构建随机生存森林
rsf_t <- rfsrc(Surv(futime,fustat)~.,data = rt,
ntree = 1000,nodesize = 15,##该值建议多调整
splitrule = 'logrank',
importance = T,
proximity = T,
forest = T,
seed = 123)
rsf_t
#树的棵数与错误率的曲线图
pdf("error.pdf",10,5)
plot(rsf_t)
dev.off()
library(survminer)
#KM+ROC 分析
## 获得每个样本的 riskscore,进一步进行下游分析
score_t <- data.frame(rt[,c(1,2)],Score=rsf_t$predicted)
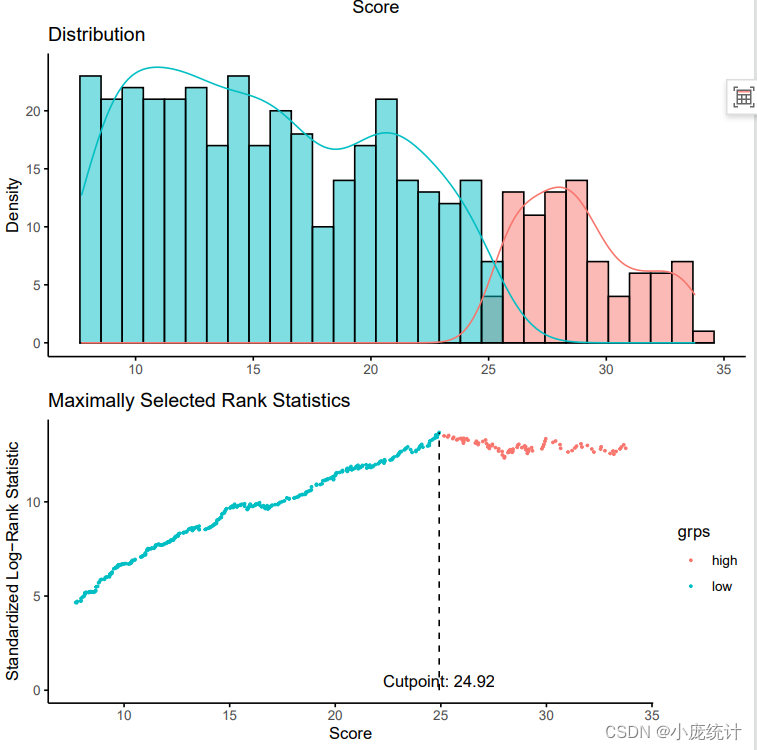
cut <- surv_cutpoint(score_t,'futime','fustat','Score')
cut
#绘制高低风险组的阈值图
plot(cut)
## 生存分析
cat <- surv_categorize(cut)
fit <- survfit(Surv(futime,fustat)~Score,cat)
#mytheme <- theme_survminer(font.legend = c(14,'plain', 'black'),
# font.x = c(14,'plain', 'black'),
# font.y = c(14,'plain', 'black')) ## 自定义主题
diff=survdiff(Surv(futime, fustat) ~Score,data = cat)
pValue=1-pchisq(diff$chisq,df=1)
if(pValue<0.001){
pValue="p<0.001"
}else{
pValue=paste0("p=",sprintf("%.03f",pValue))
}
#绘制训练集的K-M曲线
pdf("KM_train.pdf",width = 6.5,height =5.5)
ggsurvplot(fit,cat,
conf.int=T,
pval=pValue,
pval.size=6,
legend.title="Risk",
legend.labs=c("High risk", "Low risk"),
xlab="Time(years)",
break.time.by = 1,
palette=c("#ED0000FF" ,"#0099B4FF" ),
risk.table=TRUE,
risk.table.title="",
risk.table.height=.25)
dev.off()
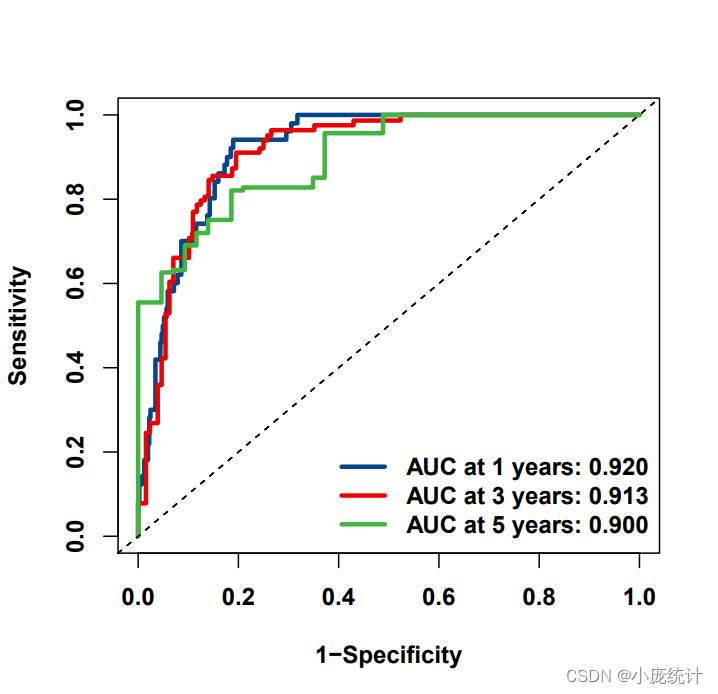
## ROC
library(timeROC)
col <- c("#0099B4FF","#925E9FFF","#FDAF91FF") ## 自定义颜色
ROC_rt <- timeROC(score_t$futime,score_t$fustat,score_t$Score,
cause = 1,weighting = 'marginal',
times=c(1,3,5), ROC=TRUE)#,iid = T
ROC_rt
#绘制训练集的ROC曲线
pdf("ROC_train.pdf",5,5)
par(cex.axis=1,cex.lab=1,font.axis=2,font.lab=2)
plot(ROC_rt,time=1,col="#00468BFF",title=FALSE,lwd=3)
plot(ROC_rt,time=3,col="#ED0000FF",add=TRUE,title=FALSE,lwd=3)
plot(ROC_rt,time=5,col="#42B540FF",add=TRUE,title=FALSE,lwd=3)
legend('bottomright',
c(paste0('AUC at 1 years: ',sprintf("%.03f",ROC_rt$AUC[1])),
paste0('AUC at 3 years: ',sprintf("%.03f",ROC_rt$AUC[2])),
paste0('AUC at 5 years: ',sprintf("%.03f",ROC_rt$AUC[3]))),
col=c("#00468BFF", "#ED0000FF", "#42B540FF"),lwd=3,bty = 'n',text.font=2)
dev.off()
## 测试集
testdata=read.table(file = "testdata.txt", header=T, sep="\t", row.names=1,check.names=F)
testdata$futime=testdata$futime/12
rsf_v<- predict(rsf_t,newdata = testdata,proximity=T)
score_v <- data.frame(testdata[,c(1,2)],Score=rsf_v$predicted)
#将测试集根据cutoff值划分为高风险组和低风险组
score_v$Group <- ifelse(score_v$Score>cut$cutpoint$cutpoint,'high','low')
fit <- survfit(Surv(futime,fustat)~Group,score_v)
diff=survdiff(Surv(futime, fustat) ~Group,data = score_v)
pValue=1-pchisq(diff$chisq,df=1)
if(pValue<0.001){
pValue="p<0.001"
}else{
pValue=paste0("p=",sprintf("%.03f",pValue))
}
#绘制测试集K-M曲线
pdf("KM_tes.pdf",width = 6.5,height =5.5)
ggsurvplot(fit,score_v,
conf.int=T,
pval=pValue,
pval.size=6,
legend.title="Risk",
legend.labs=c("High risk", "Low risk"),
xlab="Time(years)",
break.time.by = 1,
palette=c("#ED0000FF" ,"#0099B4FF" ),
risk.table=TRUE,
risk.table.title="",
risk.table.height=.25)
dev.off()
ROC_rt <- timeROC(score_v$futime,score_v$fustat,score_v$Score,
cause = 1,weighting = 'marginal',
times=c(1,3,5), ROC=TRUE)#,iid = T
ROC_rt
#绘制测试集的ROC曲线
pdf("ROC_test.pdf",5,5)
par(cex.axis=1,cex.lab=1,font.axis=2,font.lab=2)
plot(ROC_rt,time=1,col="#00468BFF",title=FALSE,lwd=3)
plot(ROC_rt,time=3,col="#ED0000FF",add=TRUE,title=FALSE,lwd=3)
plot(ROC_rt,time=5,col="#42B540FF",add=TRUE,title=FALSE,lwd=3)
legend('bottomright',
c(paste0('AUC at 1 years: ',sprintf("%.03f",ROC_rt$AUC[1])),
paste0('AUC at 3 years: ',sprintf("%.03f",ROC_rt$AUC[2])),
paste0('AUC at 5 years: ',sprintf("%.03f",ROC_rt$AUC[3]))),
col=c("#00468BFF" ,"#ED0000FF" ,"#42B540FF"),lwd=3,bty = 'n',text.font=2)
dev.off()根据预测评分划分高低风险组
ROC曲线
K-M曲线

以上就是随机生存森林,筛选基因以及构建生存预测模型的整个过程。
值得注意的是,当我们进行第一次变量筛选的时候,可能很难达到数目很小的基因个数,对于这种情况,我们可以进行二次迭代,就是将得到的基因再次放进去进行筛选,两次迭代后,基因数目大概在5-15的时候,就可以用来建模。


















 1007
1007

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











