






深度测序(deep sequencing),下一代测序(NGS),二代测序或者短读长测序(Shotr-read sequencing)在生命科学领域日趋成熟,甚至目前也发展出了单分子实时测序的第三代测序。利用测序研究生命活动的基本规律日趋重要,转录组测序,单细胞测序等等都已经成为大规模地研究基因的手段,使得研究人员可以根据数据,利用统计学的检验进行无偏见的选取目的基因。
但是您可能会被那些令人生畏的术语、工具包括bowtie2、BWA、NGS,samtools、pysam等词所吓倒,同时还会遇到同样令人生畏的技术示例,如Illumina、焦磷酸测序、SOLiD等。但不用担心,我们为您提供帮助。
1.二代测序:从FASTA到FASTQ
现在,让我们深入研究序列数据文件,即FASTQ文件,该文件包含所有原始序列(通常成为读段,reads)。FASTQ文件可以理解为FASTA文件的增强版本。
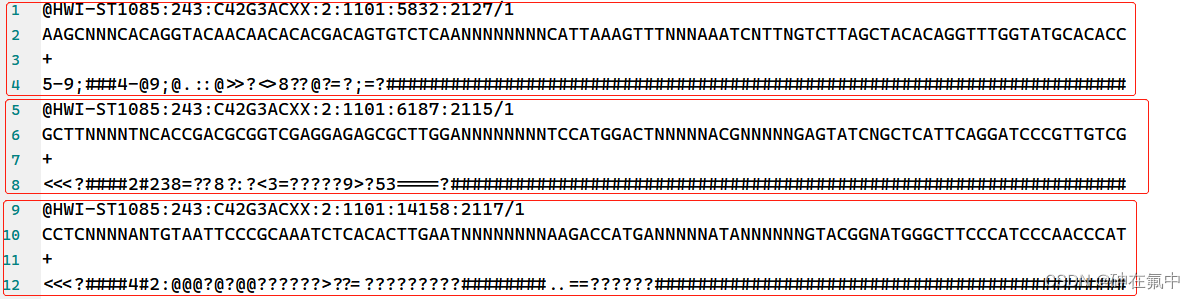
如上图所示,每个读段均由一个4行组成的段来表示。
1.第一行以符号@开头,并唯一标识读段的内容。它通常包含有关生成该reads的机器信息,以及在flow cell 上的位置等其他信息。除非reads是成对出现的,否则我们并不需要了解这些信息。这实际上与FASTA格式文件的“>”行中的信息具有相同的用途。
2.第二行包含由机器识别出的实际序列。
3.第三行以符号+开头,并将序列信息与含有的序列质量得分的下一行分开。
4.最后一行是序列质量得分;这是原始机器用来评估第二行中序列质量的得分。这些很重要,因为当将所有序列放在一起时,他们会被下游的算法所使用。二代测序之所以会有碱基质量得分,是因为它是一簇一簇测的,一簇里面有好几个由一段reads组成的相同碱基,它们在相同的时间理论上应发出相同的荧光,但总有几个reads会发出不同的荧光而导致存在误差,一般用G30表示。
理论上,我们可以编写像FASTA那样,直接编写一个Python程序来读取FASTAQ文件,但实际上FASTAQ文件很少被用来直接分析,而是将它们用作其他工具的输入。比对是NGS数据最常见的任务之一。比对就是指在FASTQ文件中获取所有的单个reads,并找到他们在给定基因组中的位置。该基因组成为参考基因组。目前由许多工具可以执行此任务,其中最常见的就是bowtie2和bwa,但是目前都只是支持Linux。下面以bowtie2为例介绍比对流程。
比对会生成一个比对文件,而bowtie2会生成一个SAM文件。这是一个未压缩的文本文件,每个reads都站一行,其中比对软件对reads所在的基因组上原始位置的“最佳猜测”。SAM文件还包含那些完全没有被比对的reads。
1.1BAM文件:查看(view)、排序(sort)和索引(index)
由于SAM文件在实践中的用于有限,且文件通常非常庞大。因此我们通常会把SAM文件转化为BAM文件,SAM文件转化为BAM文件后在文件大小方面,有了大幅的缩小,但为了BAM文件能更好地用于数据分析,还需要另外两个步骤:排序和索引。所有的这三个步骤都是通过samtools工具(http://samtools.sourceforge.net/)来完成的。
1.查看:用samtools的view命令完成从SAM到BAM的转换。
2.排序:排序就是将所有靠近染色体末端的读段都放在文件的开头。这样可以快速的在特定区域中找到所有reads,因为它们在文件中彼此相连。这可以通过samtools的sort命令来实现。
3.索引:索引必须始终在排序之后进行,它会创建一个额外的BAI文件。该文件基本上相当于一个查找表,以便可以通过染色体名称轻松地检索reads,而无需从头到尾搜索整个文件。这可以通过samtools命令中的index实现。
让我们以流程图的形式展示此过程:
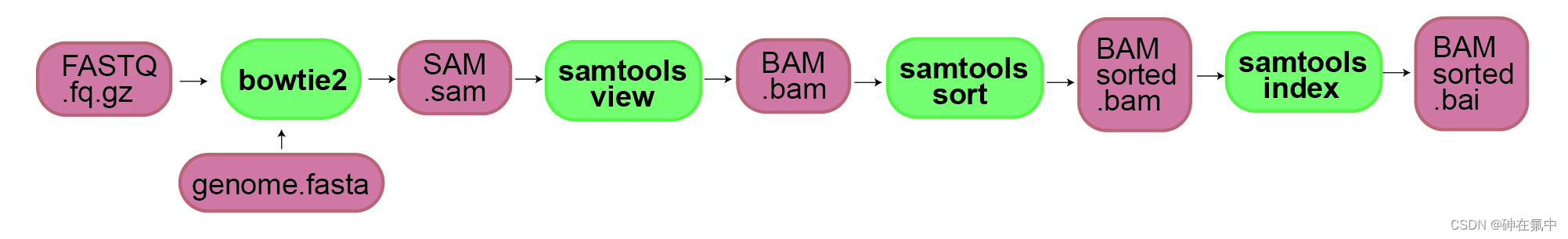
有关samtools和bowtie2的更多信息请分别访问samtools.sourceforge.net和bowtie-bio.sourceforge.net。
现在让我们停下来盘点一下。您可能已经注意到,为了完成比对这一小小的过程,你需要运行许多不同的软件和步骤,且有些步骤还耗时较长,要等上一步完成后在进行下一步。您可能会认为将SAM转化为BAM,排序和索引都内置到bowtie2中会更好,这样就可以节省很多时间。也许您是对的,但是就像许多软件一样,NGS工具已经发展出了一种将不同的软件连接在一起的“拼盘”方式。这就需要用到Python,我们可以利用Python的subprocess模块(https://docs.python.org/3/library/subprocess.html)来调用shell命令,以包装(wrap)或者说是封装(encapsulate)其中许多复杂的部件以便更利于重用流程。下面我们将以苹果的单末端转录组的数据来演示如何用subprocess模块实现不同软件的包装。
2.subprocess模块实现比对,SAM转BAM,排序,索引全部流程
2.1.创建python的test环境
先激活base环境,然后输入命令:
conda create -n test python=3.82.2. 激活test环境
Test环境安装好后,输入命令:
conda activate test2.3.bowtie2,samtools,pysam的安装
利用conda安装bowtie2,输入命令:
conda install bowtie2利用conda安装samtools,输入命令:
conda install samtools利用pip安装pysam,输入命令:
pip install pysam -i https://pypi.tuna.tsinghua.edu.cn/simple2.4.建立工作目录
先建立一个python_RNA-seq的文件夹,然后在python_RNA-seq文件夹下建立以下4个文件夹(blast_result[存储比对结果SAM文件],genome[存储基因组的索引文件],mapping[代码文件夹,之后的.py文件就放在这],sequence_data[存储测序的reads文件])。
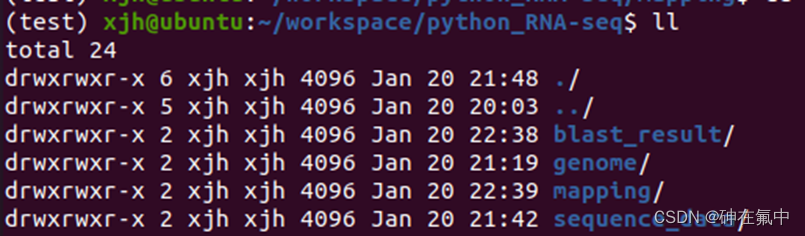
图1 目录样式
sequence_data文件夹下的测序文件(.fq.gz)一定要以样品名称命名,有生物学重复的用1,2,3表示。注意这里为了方便大家运行,我们对测序得到的reads文件做了相关处理,文件大小从原来的1G多删为40M,这样就方便在自己的电脑虚拟机上也可正常运行,文件的获取方式我们放在了最后。且一般的项目都会有多个样品,因此我们也用了6个样品,如图2所示:

图2 sequence_data文件夹下的目录样式
在mapping文件夹下存放基因组文件(genome.fasta)和python代码(mapping.py)。
2.5.构建参考基因组的索引
在python_RNA-seq目录下输入命令:
bowtie2-build ./mapping/genome.fasta ./genome/genome_index
红色表示基因组文件所在的位置
绿色表示建立的索引的输出位置,位置在genome文件夹下,且文件都以genome_index命名。

图3 构建基因组索引的样式,图片上的代码少了后面的输出索引文件的命名:genome_index
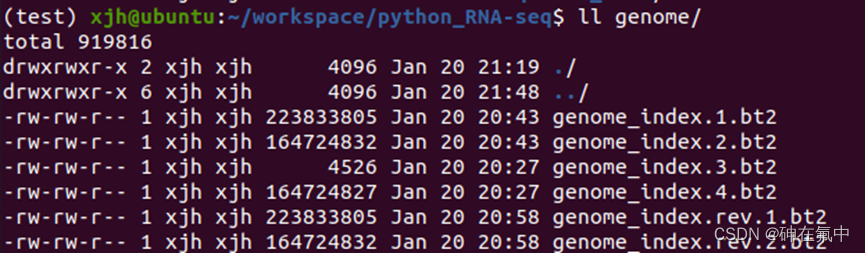
图4 建立基因组索引后输出结果的样式,都是genome_index命名,若不以genome_index则需mv命令手动更改,不然后续的python代码都要改
2.6.比对,SAM转BAM,排序,索引
进入mapping 文件夹,将mapping.py文件放入mapping文件夹下,然后输入命令:python mapping.py 运行python文件即可。以下是mapping.py的代码,代码中标了注释,并在下面会详细说明以方便大家的理解。
import subprocess
import os
import pysam
#利用pysam,构建排序和索引的函数
def sort_and_index(bam_filename):
#os.path.splitext分别读取文件的前缀(样本名称,prefix)和后缀(suffix)
prefix,suffix = os.path.splitext(bam_filename)
#字符串拼接,产生排序好的文件名称(样品名称+.sorted.bam)
sorted_bamfilename = prefix + '.sorted.bam'
print('generated resorted BAM:',sorted_bamfilename)
#进行排序
pysam.sort('-o',sorted_bamfilename,bam_filename)
print('index BAM:',sorted_bamfilename)
#建立索引
pysam.index(sorted_bamfilename)
return sorted_bamfilename
#file_number记录要读取文件的个数
file_number=0
#file_names为存储测序样品名称的列表
file_names = []
#使用os模块,读取测序得到的.fq.gz文件,最后将该目录下的所有样品的文件名存储到file_names中
for root,dirs,files in os.walk(r"/home/xjh/workspace/python_RNA-seq/sequence_data"):
for file in files:
if os.path.splitext(file)[1]==".gz":
file_number+=1
file_names.append(file[:10])
#打印读取到的文件个数,以方便检查一下是否所有文件都已经读取
print('the number of sequence data is ',file_number)
#for循环遍历存储样本名称的列表(file_names)
for i in file_names:
#bowtie2 比对到参考基因组,得到SAM文件
with open('{}.sam'.format(i), 'w') as output_sam:
subprocess.check_call('bowtie2 -p 2 -x ../genome/genome_index -U ../sequence_data/{}.fq.gz -S ../blast_result/{}.sam 1> ../blast_result/{}.log 2>&1'.format(i,i,i), shell=True,stdout=output_sam)
#用samtools工具将SAM转化为BAM文件
with open('{}.bam'.format(i),'w') as output_bam:
subprocess.check_call(['samtools', 'view', '-b', '-S','../blast_result/{}.sam'.format(i)],stdout = output_bam)
#排序并建立索引,以方便IGV可视化
output_sorted_bamfilename = sort_and_index('{}.bam'.format(i))2.6.1.python代码中比对这一步shell命令的解释
'bowtie2 -p 2 -x ../genome/genome_index -U ../sequence_data/{}.fq.gz -S ../blast_result/{}.sam 1> ../blast_result/{}.log 2>&1'.format(i,i,i)
红色表示第五步基因组索引的文件位置
绿色表示所有的测序文件(.fq.gz)所在的位置,-U是但末端,若是双末端测序,则是-1 文件1 -2 文件2
蓝色表示比对后的结果(SAM文件)的输出位置
黄色表示比对过程中的日志文件(LOG文件)的输出位置
2.6.2.python代码中SAM文件转化为BAM文件这一步shell命令的解释
['samtools', 'view', '-b', '-S','../blast_result/{}.sam'.format(i)]
红色表示上一步比对结果SAM文件所在的位置
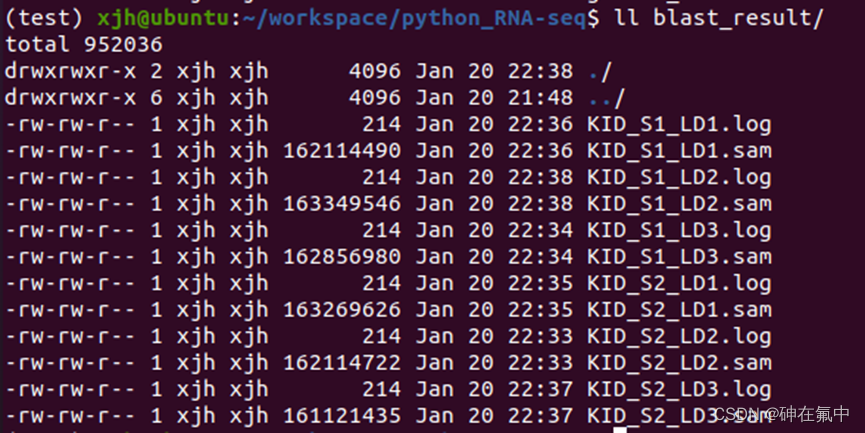
图5 比对的输出结果所在的位置
2.7.输出结果展示
若代码正常运行,则会出现如下图所示的样子。

图6 代码正常运行的样子
最后排序和索引的结果会在mapping文件夹中。
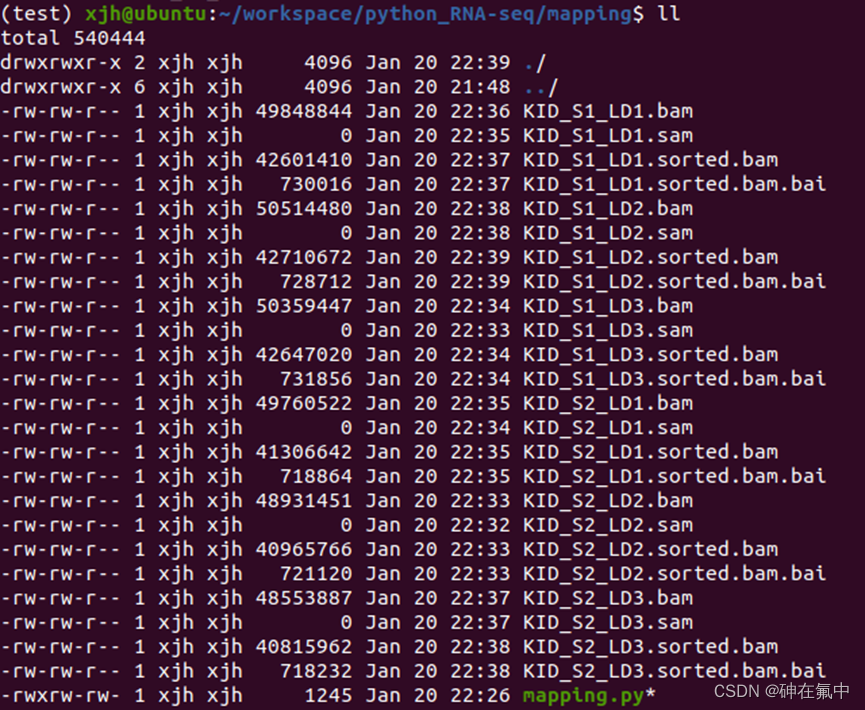 图7 最后的排序和索引的输出结果。
图7 最后的排序和索引的输出结果。
PS:上述代码运行所需要的文件
链接: 百度网盘 请输入提取码
提取码: rysd















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









