






在生信分析中,由于论文中提供的数据有限,很多时候需要自己下载论文中的测序数据重新进行分析,从而得到自己想要的数据。而论文中的作者往往会把测序数据上传到NCBI中。因此学会如何从NCBI下载测序数据非常重要。步骤如下:
1.查找文献中的序列号
一般在文献的材料和方法里面或者在文献的末尾可以找到这个序列号,例如下面这篇论文就在材料和方法中说明了自己的测序数据放在了NCBI的GEO数据库上(图1)。然后就打开NCBI的GEO数据库的网址(https://www.ncbi.nlm.nih.gov/geo/)对GSE153159这个编号进行检索(图2)。

图1 文献中序列号的查找
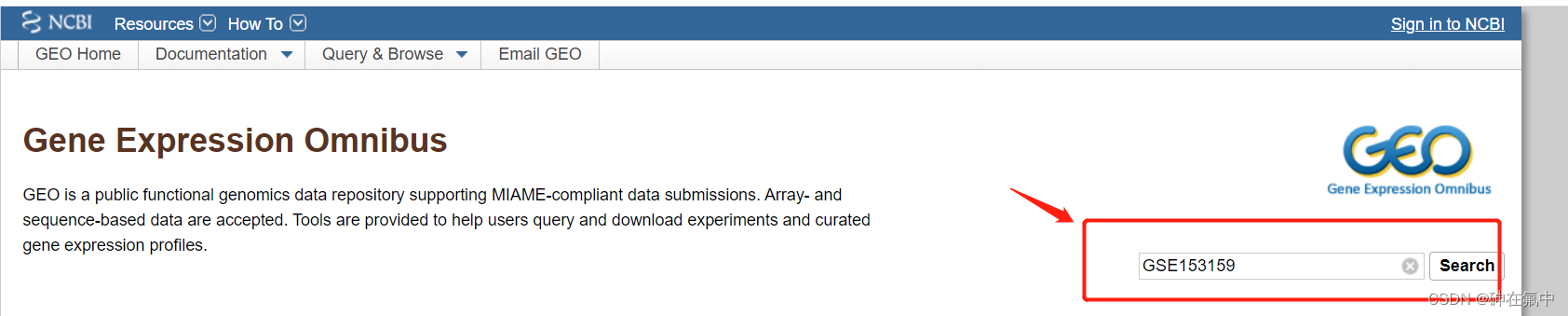
图2 对序列号进行搜索
搜索后的页面如图3所示,按照图中的操作下载自己感兴趣的样本的SRA编号,然后上传到服务器上。
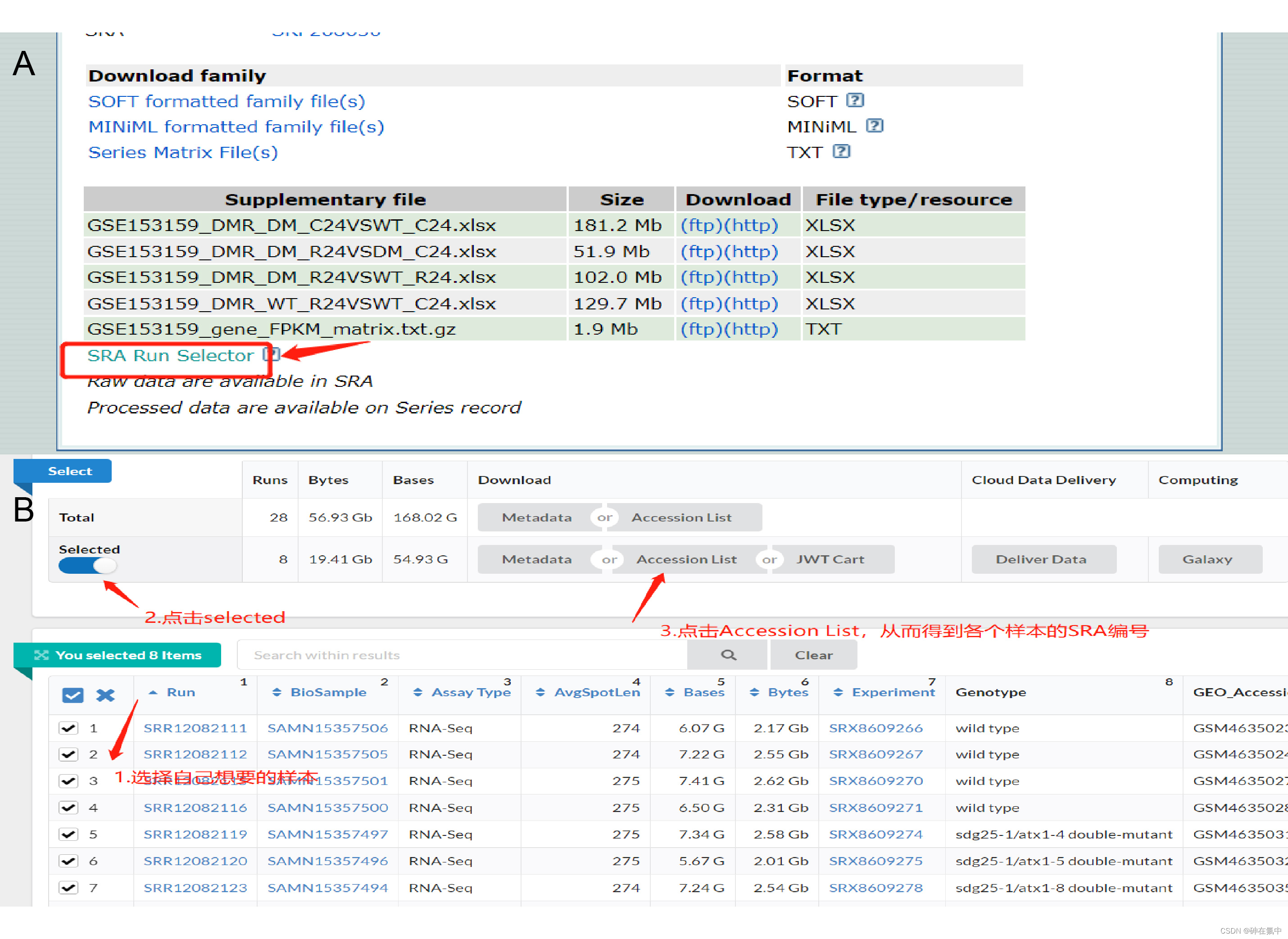
图3 (A)序列号在GEO数据库中的搜索结果 (B) 点击Accession list,下载各个样本SRA的序列号
2. FileZilla传输下载好的SRR_Acc_List.txt文件到Linux
通过FileZilla将下载好的.txt文件传输到Linux里自己的工作目录下,然后用vi编辑器按照以下格式(图4),加入样本和分组信息,用制表符分割。最后可以用cat命令查看文件的内容。
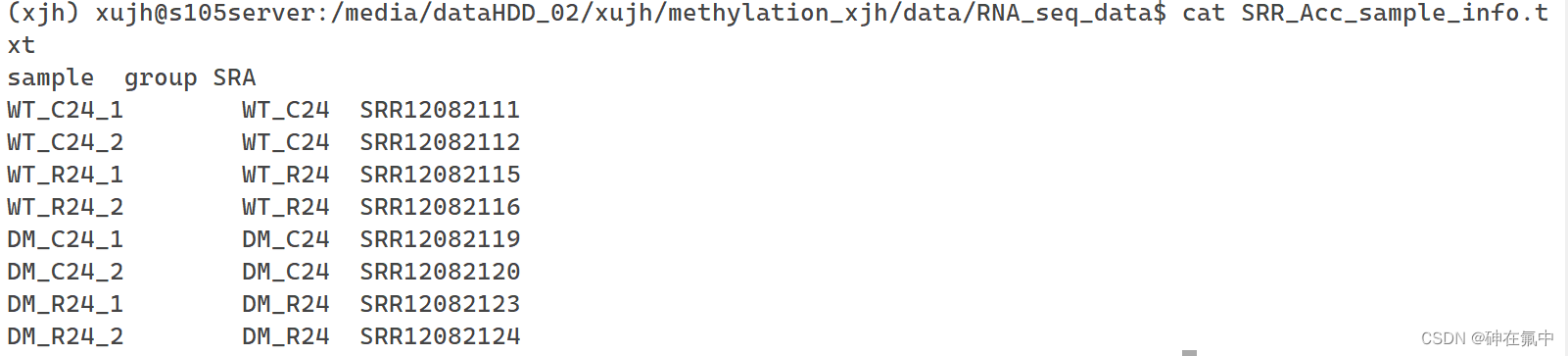
图4 将SRR_Acc_List.txt修改后的样式,文件的名称为SRR_Acc_sample_info.txt
3.Linux 安装sra-tools
在下载数据前,我们还需要在Linux下用conda下载sra-tools工具,下载命令如下:
conda install sra-tools4.测序数据下载
用awk命令一键生成代码,并且重定向到run_prefetch.sh的文件中具体代码如下:
awk '{print "prefetch "$3" &"}' SRR_Acc_sample_info.txt | grep "SRR" > run_prefetch.sh
#运行run_prefetch.sh文件
nohup sh run_prefetch.sh5 sra格式转化为fasta格式
待所有的sra文件下载完成后,用awk命名一键生成sra转化为fasta文件的代码:
awk '{print "fastq-dump --gzip --split-3 /media/dataHDD_02/xujh/methylation_xjh/data/RNA_seq_data/"$2"/"$2".sra &"}' run_prefetch.sh > transform_sra_to_fasta.sh
#运行transform_sra_to_fasta.sh
nohup sh transform_sra_to_fasta.sh代码中的参数--gzip一定要加上,不然转化出来的是没有压缩的fasta文件,不加的话会导致文件内存很大。

















 3747
3747

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









