










基于配体的药物设计(LBDD)
基本原理
基于配体的药物设计(Ligand-based Drug Design,LBDD)是药物研发中的一种方法,主要依赖于已知的配体(通常是具有活性的小分子化合物或药物)来指导新药的发现与设计。这种方法利用已知配体的结构信息,通过比较、模拟或改造等策略来设计新的候选药物分子。基于配体的设计通常不需要靶标的三维结构信息,而是通过分析配体与靶标之间的相互作用来推测潜在的药物活性。这一类设计方法主要包括定量构效关系模型、药效团模型以及相似性搜索方法。
常见方法
定量构效关系模型
定量构效关系(Quantitative Structure-Activity Relationship,QSAR)是指利用理论计算和统计分析工具来研究一系列化合物的结构(包括三维分子结构和电子结构)与其生物效应(如药物的活性、毒性、药效学性质、药代动力学参数和生物利用度等)之间的定量关系。QSAR可以帮助快速筛选候选分子,优化分子的活性、选择性和毒性。该方法主要包括二维构效关系(2D-QSAR)和三维构效关系(3D-QSAR)。
- 2D-QSAR方法如Hansch模型和Free-Wilson模型等,但由于它们不能细致地反映分子的三维构造与生理活性之间的关系,因此逐渐被3D-QSAR方法所取代。
- 3D-QSAR方法以配体和靶点的三维构造特性为基础,根据分子的内能变化和分子间相互作用的能量变化,将已知一系列药物的理化参数和三维构造参数与药效拟合出定量关系,再以此关系预测新化合物的活性,进行结构的优化和改造。
在3D-QSAR方法中,比较分子场分析法(CoMFA)和比较分子相似因子分析法(CoMSIA)是两种常用的方法。它们通过分析药物分子周围的作用势场分布,把势场与药物分子的生物活性定量地联系起来,用以推测靶点的某些性质,并可依此建立作用模型来设计新的化合物,定量地预测其活性强度。
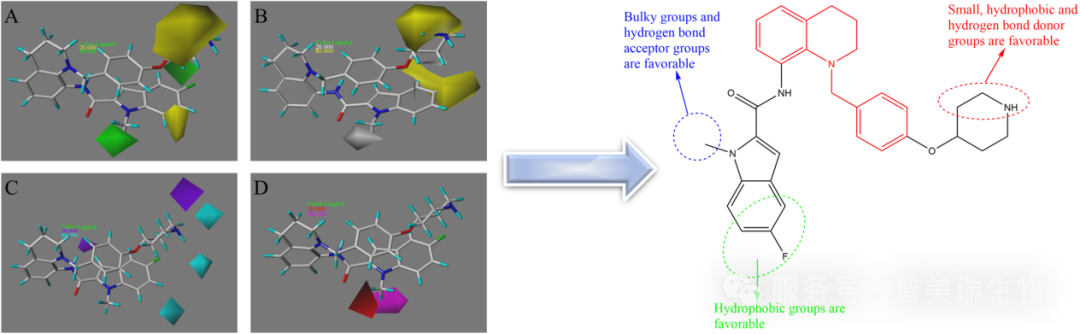
图 1. 基于CoMSIA方法得到四氢喹啉类衍生物的构效关系信息[1]
药效团模型
药效团模型是一种基于已知活性配体的三维结构特征,识别对靶标活性至关重要的化学基团(可以与受体结合位点形成氢键、静电、疏水等非键相互作用的原子或原子团),并生成一个空间结构模型。药效团模型反映了配体与靶标相互作用的核心化学特性。
建好药效团模型后,可利用药效团从已知小分子数据库中寻找新骨架活性分子,也可以定性或定量地解释化合物构效关系(QSAR),阐明化合物的选择性机理。由于药效团模型中包含了药物和靶点结合的三维结构信息,因此根据药效团模型不仅能优化化合物的结构,还能设计出新的先导化合物。
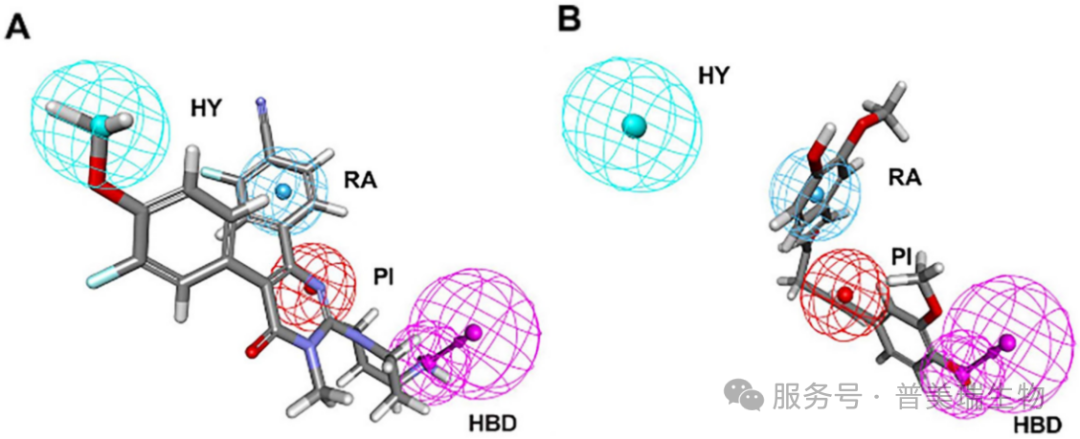
图 2. LSD1抑制剂的药效团模型[2]
相似性搜索
分子相似性是指两个分子在结构或性质上相似的程度,包括了分子的物理性质、结构、形状、三维分子场等方面的相似性。相似性原理指出,结构类似的化合物具有相似的物化性质和生理活性,通过分析已知活性配体的结构特征,寻找与之结构相似的分子,这些相似分子可能具有类似的生物学效应。
相似性搜索主要有以下三类:
1. 子结构搜寻
子结构搜寻是指在大型化学数据库中查找包含特定子结构的化合物。这些子结构通常是与已知活性化合物共有的关键药效基团或功能部分。通过子结构搜寻,研究人员可以快速缩小筛选范围,专注于那些可能具有相似生物活性的化合物,从而加速药物发现过程。
2. 基于分子指纹的相似性搜索
分子指纹是一种将化合物结构信息转化为一系列二进制代码(即0和1)的方法,这些代码代表了化合物中特定子结构或特征的存在与否。通过比较不同化合物的分子指纹,可以评估它们之间的相似性,并预测新化合物的潜在活性。
3. 基于分子形状的相似性搜索
基于分子形状的相似性搜索,通过评估分子间的三维重叠来识别可能具有相似性质的候选化合物。如果两个分子具有相似的形状,它们可能具有相似的性质,从而有可能与相同的受体结合并产生相似的生物活性。
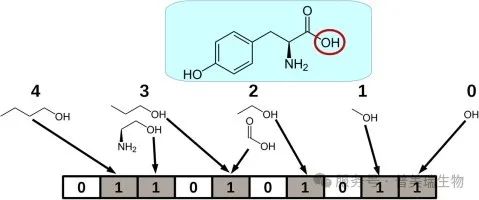
图 3. 酪氨酸结构的分子指纹表示[3]
小结
基于配体的药物设计是一种重要的药物发现策略,尤其适用于没有靶标三维结构的情况。通过对已知配体的结构、活性和相互作用特征进行分析,研究人员可以设计出新的药物候选分子,节省了大量的实验时间和成本。不过,近年来,随着结构解析技术(如X射线晶体学、核磁共振成像NMR、冷冻电镜Cryo-EM)和AlphaFold的迅速发展,越来越多的蛋白质三维结构被解析,这极大地推动了基于结构的药物设计的发展。
参考文献
- Xu Y, Fan B, Gao Y, et al. Design two novel tetrahydroquinoline derivatives against anticancer target LSD1 with 3D-QSAR model and molecular simulation[J]. Molecules, 2022, 27(23): 8358.
- Yin Z, Liu S, Yang X, et al. LSD1-Based Reversible Inhibitors Virtual Screening and Binding Mechanism Computational Study[J]. Molecules, 2023, 28(14): 5315.
- Cereto-Massagué A, Ojeda M J, Valls C, et al. Molecular fingerprint similarity search in virtual screening[J]. Methods, 2015, 71: 58-63.

















 1823
1823

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









