










原创 普美瑞生物科技 普美瑞生物 2024年11月26日 14:46 江苏
分子对接(Molecular Docking)是分子模拟研究中的重要方法之一,旨在预测小分子与靶蛋白之间的结合模式及相互作用,广泛应用于药物发现和设计。近年来,随着计算能力的提升以及机器学习算法的发展,分子对接的精确度和效率显著提高,进一步巩固了其在现代药物研发中的重要地位。
1.分子对接的基本原理
分子对接的关键步骤包括预测小分子与靶蛋白的结合构型、结构优化及通过评分函数评估结合亲和力。
结合模式的预测主要基于锁钥模型和诱导契合模型(图1)。在对接过程中,假设靶蛋白的构象保持不变,仅调整配体构象的对接方式称为刚性对接;而允许靶蛋白根据配体的变化调整构象的对接方式称为柔性对接。此外,还有允许靶蛋白活性位点附近的部分残基进行构象调整的半柔性对接方法。
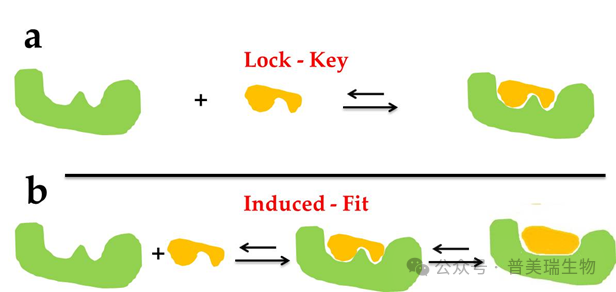
图1. 锁钥模型与诱导契合模型
在确定靶蛋白活性位点附近的可结合空间后,使用特定的搜索算法在空间内寻找能量最低或亲和力最高的配体构象。搜索算法可系统分类为三大类:系统方法(增量构造、构象搜索、数据库),随机方法(蒙特卡罗、遗传算法),模拟方法(分子动力学,能量最小化)[1,2]。
不同的搜索算法在计算精度和运行效率上各不相同。系统方法通过系统化的搜索策略逐步生成配体构型,能够全面探索所有可能的构象,减少遗漏,但对于高自由度分子或大分子体系,计算量巨大且容易陷入局部最优,难以找到全局最优。随机方法能够探索复杂的能量表面,避免局部最优,找到全局最优的可能性较大,但由于随机性,可能导致结果不一致,难以复现。模拟方法则能够考虑分子之间的真实相互作用、动态行为以及溶剂效应,提供更准确的结合预测,但计算资源耗费巨大,且对初始构象敏感,容易陷入局部最小值。
在获得可能的配体与靶蛋白结合方式后,需要使用打分函数对预测结果进行评估。根据理论基础和计算方式,打分函数可分为经验型、基于力场和基于知识三种[1,2]。经验型打分函数通过拟合实验数据,计算速度快,适合大规模筛选,但普适性差且依赖数据的准确性。基于力场的打分函数能够准确反映分子间的物理相互作用,适用于复杂体系,但计算量大且常忽略溶剂效应。基于知识的打分函数利用统计数据进行预测,适合处理已有数据丰富的常见系统,但在处理新结构时准确性不足。
搜索算法和打分函数在分子对接中均扮演着至关重要的角色。搜索算法负责在庞大的配体空间中高效寻找可能的结合构型,确保探索足够的空间以发现最佳结合位点。打分函数用于评估这些构型的结合质量,提供配体与靶蛋白相互作用的定量信息。两者相辅相成,搜索算法的效率和全面性决定了可评估的构型数量,打分函数的准确性和可靠性直接影响最终的预测结果。因此,优化搜索算法和打分函数是提高分子对接成功率和准确性的关键。
2. Autodock Vina
AutoDock是由斯克里普斯研究所开发的分子对接程序,自推出以来已成为分子对接领域广泛应用的强大工具。据统计,2015至2020年期间,约40%的相关研究使用AutoDock进行分子对接[3](图2)。
AutoDock Vina基于AutoDock,针对打分函数和搜索算法进行了优化,显著提升了准确性和运行效率。相比AutoDock,Vina的运行速度大幅提高,同时也更好地处理了分子间的相互作用,进一步增强了对复杂体系的适应能力(图3)。Vina在大规模虚拟筛选中的表现尤为突出,为加速药物研发提供了强有力的支持。
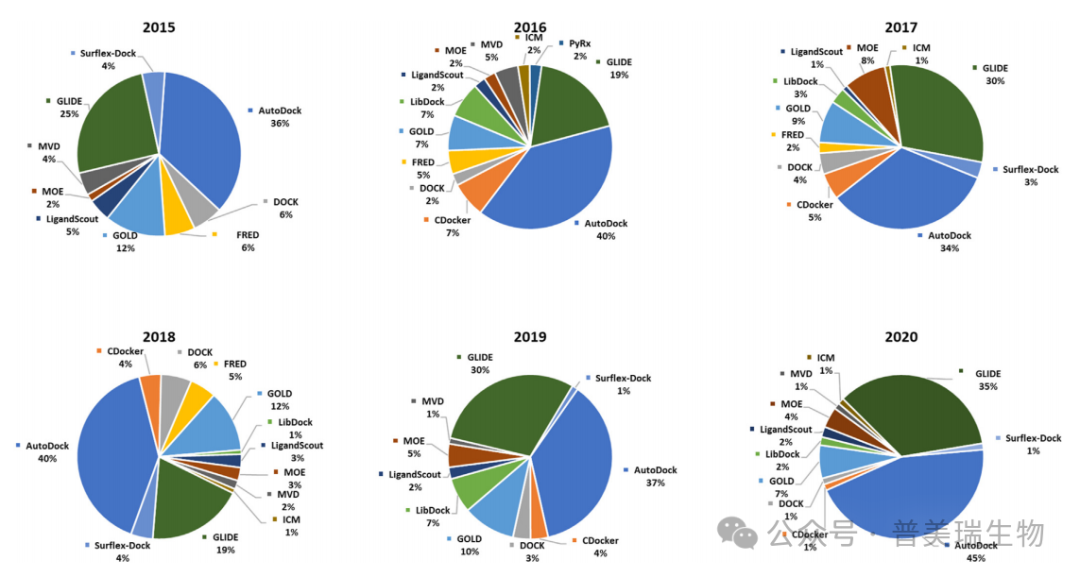
图2. 2015~2020年间AutoDock与其他对接程序在分子对接研究中使用占比
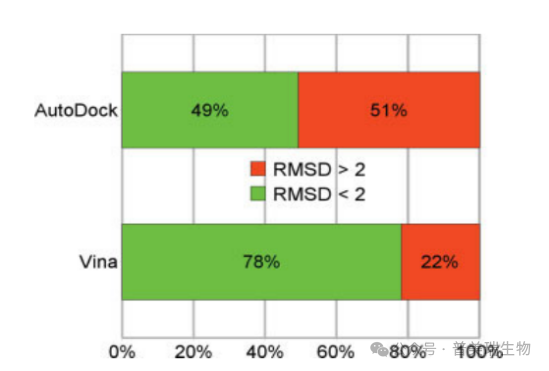
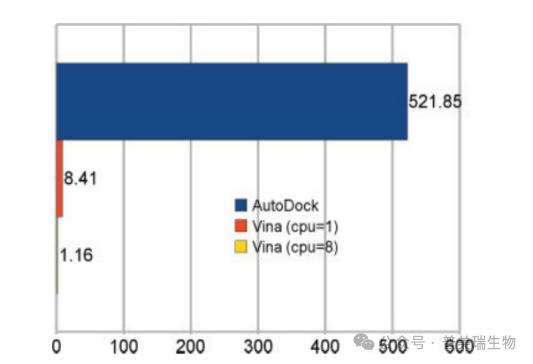
图3 AutoDock与AutoDock Vina的预测准确度(上)与预测时间(下)对比
2.1 Autodock Vina的搜索算法
Vina在发展过程中采用了多种随机全局搜索优化方法,包括遗传算法、粒子群优化法和模拟退火法。在2010年发布的Vina版本中,采用了Broyden-Fletcher-Goldfarb-Shanno(BFGS)方法进行局部优化。这是一种高效的拟牛顿法,利用评分函数的值及其梯度,使其在搜索过程中更快收敛到局部最优。与其他优化方法相比,BFGS在迭代时利用前一步的梯度信息,从而减少了计算时间,提高了程序的运行效率。
2.2 Autodock Vina的打分函数
Autdock Vina的打分函数形式为:
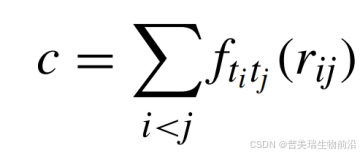
其中,ftitj是关于原子类型ti和tj的相互作用函数,ri和rj是原子i和j之间的距离。相互作用函数ftitj根据原子类型和距离的不同,定义了多种相互作用类型,包括高斯势能、排斥势能、疏水相互作用和氢键相互作用。不同的作用类型被赋予了相应的权重,并采用大量实验数据和机器学习方法优化参数模型,从而能够更全面地描述原子和官能团之间的相互作用。
3. CoDock-Ligand方法
CoDock-Ligand是一种结合模板建模与卷积神经网络(CNN)评分功能(GNINA)的分子对接方法,由常珊和孔韧团队开发。该对接方法在CASP15的高难度配体结合模式的预测中取得了第一名(图4),证明了该方法具有很好的预测效果,并且能良好的处理复杂配体结构与靶蛋白口袋之间的相互作用。
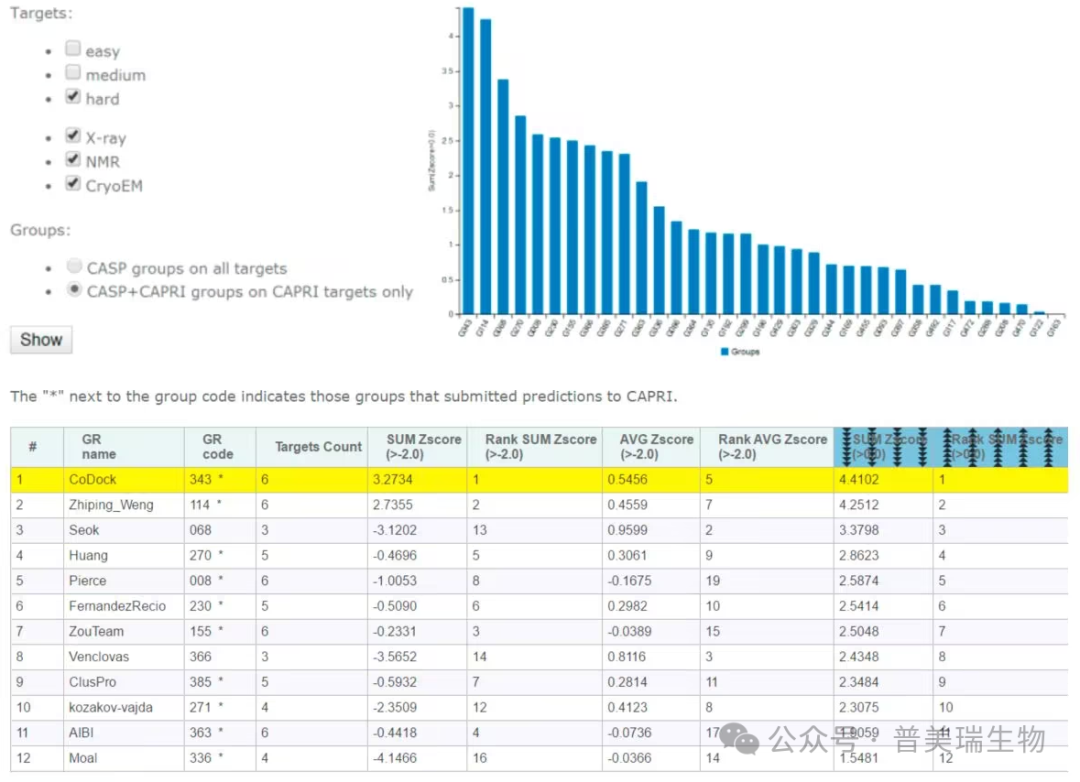
图4. CASP15困难难度的配体-靶蛋白结合预测成绩
在CASP15提供的预测体系中,CoDock-Ligand方法的预测成功率高于AutoDock Vina以及AutoDock Vina结合GNINA的评分方法[4](图5)。
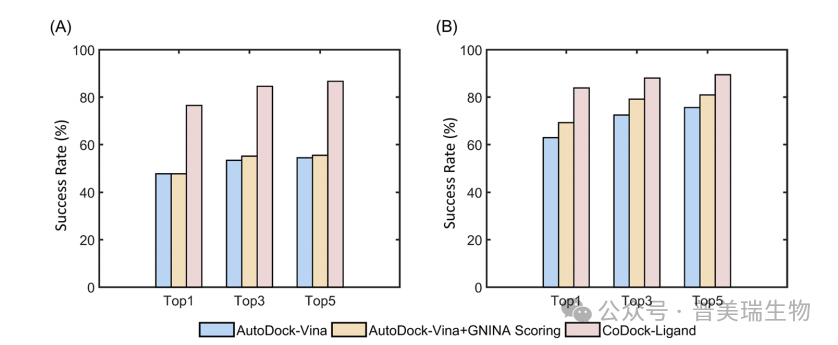
图5 CoDock、AutoDock Vina和AutoDock Vina结合GNINA的打分方法的预测成功率
3.1 CoDock的搜索算法
CoDock-Ligand开发团队开发了一种基于结构的3D对齐算法,该算法用于靶蛋白结合口袋的模板搜索和配体比对。通过这一3D对齐算法,将靶蛋白结合口袋附近的Cα原子作为节点进行建模,从而在蛋白质数据库中检索与目标蛋白质相似的模板(图6)。该过程通过将查询结构和模板结构中的原子配对,形成节点间的距离矩阵,以计算结构的相似性。此外,检索过程结合了贪心算法(Greedy Algorithm)寻找阶段性最优解,相较于传统的穷举法,具有更高的运行效率。
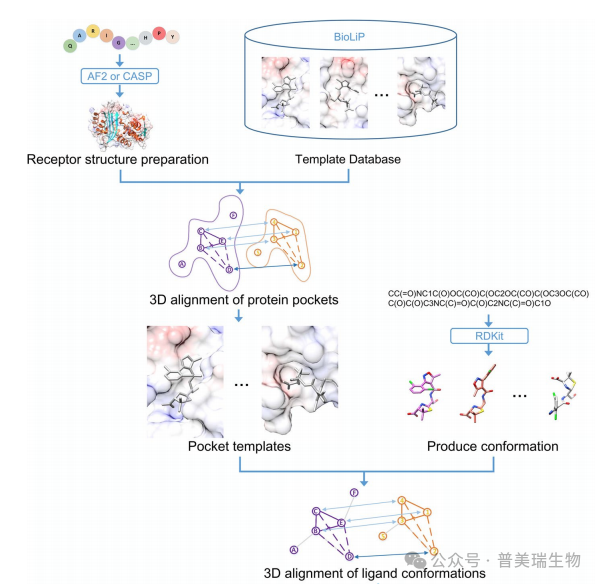
图6. CoDock-Ligand的搜索方法流程图
3.2 CoDock-Ligand方法的打分函数
CoDock-Ligand的打分函数部分使用了基于卷积神经网络(CNN)的GNINA打分功能来评估配体与受体的相互作用。GNINA是一个基于CNN的深度学习模型,能够根据空间和原子类型信息预测配体与蛋白质结合的姿态和亲和力。它将配体和蛋白质的原子类型表示为3D网格数据,并将不同原子类型的空间分布作为CNN的输入进行学习。
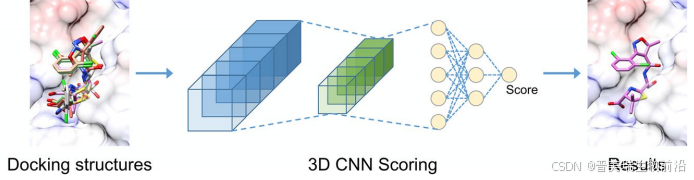 图7. CoDock-Ligand使用GNINA对预测配体构型进行打分评估流程图
图7. CoDock-Ligand使用GNINA对预测配体构型进行打分评估流程图
GNINA打分模型在晶体结构和复合对接数据集上进行训练,能够同时预测配体的对接姿态打分和结合亲和力。该打分功能可以处理较大的数据网格,确保包含配体可能结合的所有位点。同时,它可以根据打分结果对这些姿态进行排序,从而选择出最可能的结合模式。
4.使用CoDock-Ligand方法对7VU6中配体进行复原对接
在这里,我们以SARS-CoV-2 3CL蛋白酶与配体的结合晶体结构(PDB ID:7VU6)为例,使用CoDock-Ligand方法对7VU6晶体结构中的配体进行复合对接。具体操作流程如下:
①首先通过普美瑞生物官方网站(https://pumeirui.com/)中的蛋白-小分子对接模块访问CoDock-Ligand方法对接功能:
②点击“立即运行”启动蛋白-小分子对接模块
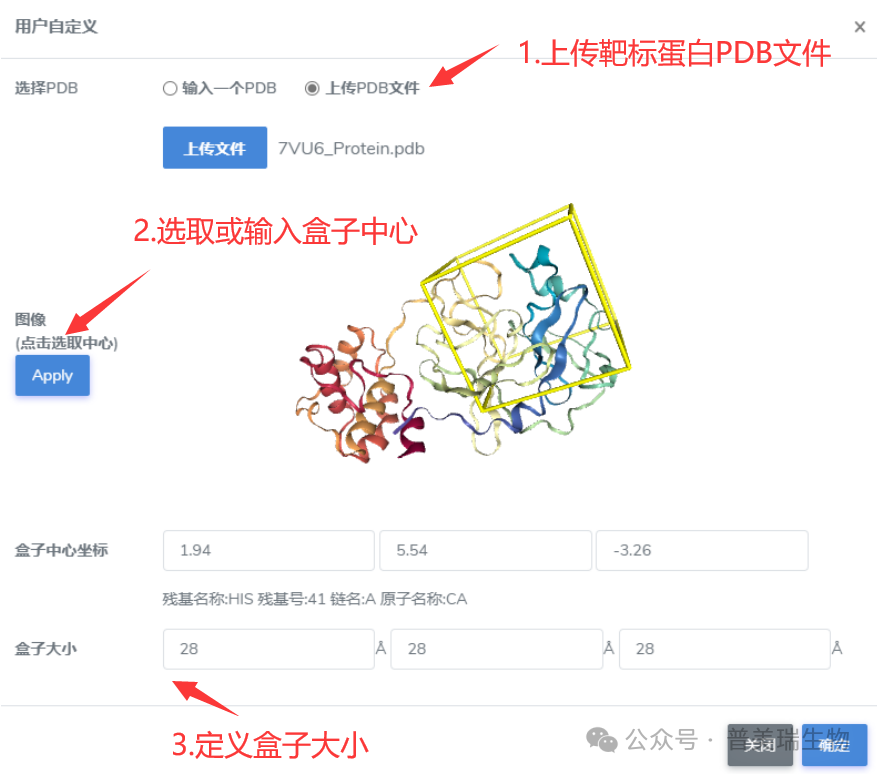
③在表单中提交靶标蛋白的信息:定义搜索空间中心、搜索空间大小、选择对接算法(选择PsDock)
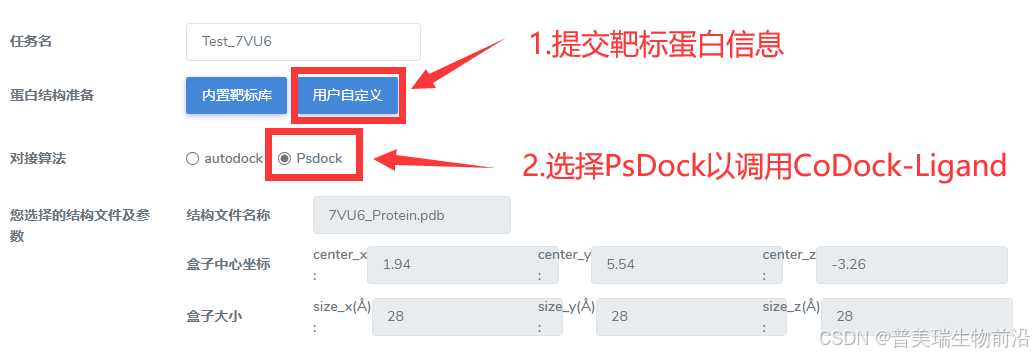 靶标蛋白信息填写方式:
靶标蛋白信息填写方式:
④输入配体信息:CoDock-Ligand提供了3种提交分子信息的方式,分别为:绘制配体分子2D结构、输入配体分子SMILES或直接提交配体分子的结构坐标文件(可接受.smi、.sdf和.mol2文件)
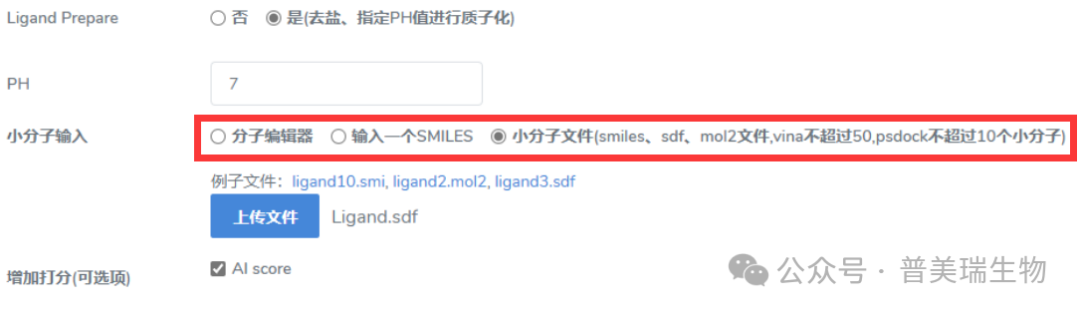
⑤点击提交键提交:

⑥运行结束后可点击“下载报告”获得对接结果,也可以使用网页内可视化界面查看对接结果
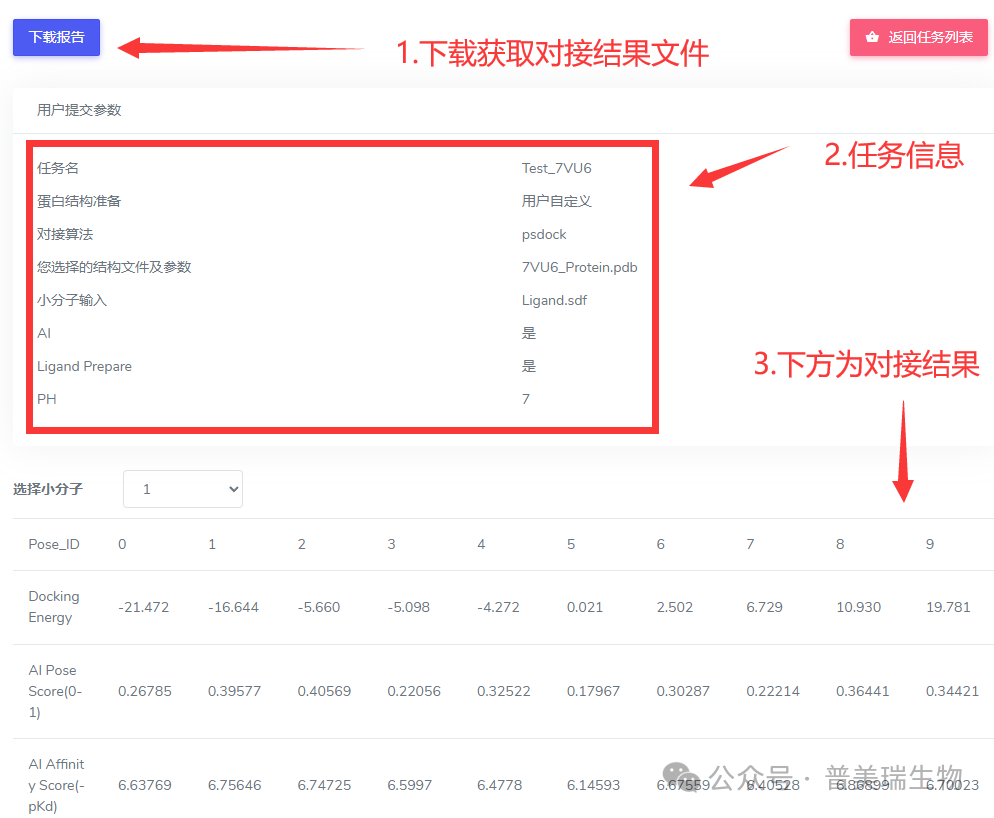
运行所得对接结果如下:
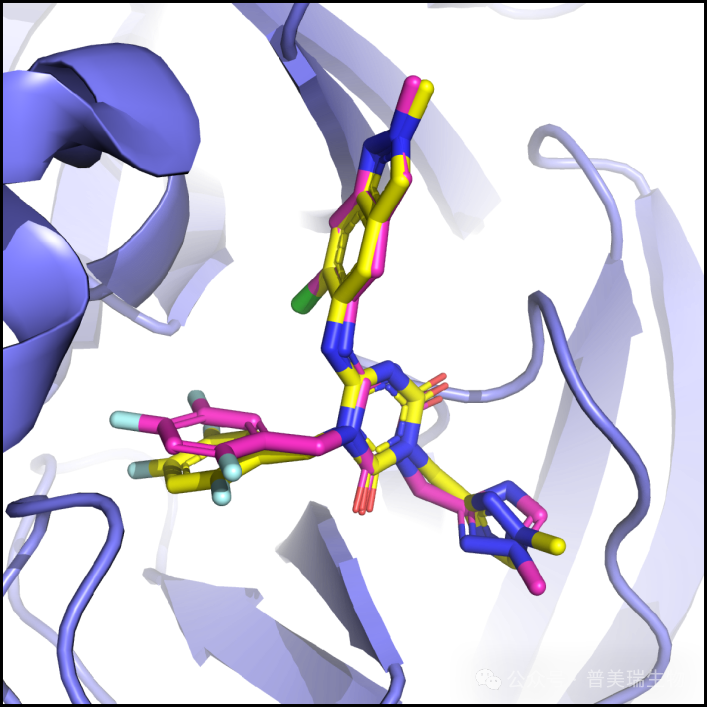
图8.使用CoDock-Ligand算法对7VU6晶体结构中的配体分子进行复原对接的结果
打开结果文件夹中的MOL_1/0.sdf,并将其与7VU6中的配体分子进行比对。黄色表示7VU6晶体结构中配体的构象,而紫色表示CoDock-Ligand方法计算的对接结果。可以观察到,CoDock-Ligand方法精确预测了配体的构象,配体中的官能团延伸方向及相对位置与晶体结构中的配体几乎一致。说明CoDock-Ligand方法能良好的预测配体分子的结合姿势。
参考文献
[1]Kitchen, D. B., Decornez, H., Furr, J. R., & Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: methods and applications. Nature reviews. Drug discovery, 3(11), 935–949. https://doi.org/10.1038/nrd1549
[2]Dias, R., & de Azevedo, W. F., Jr (2008). Molecular docking algorithms. Current drug targets, 9(12), 1040–1047. https://doi.org/10.2174/138945008786949432
[3]Sabe, V. T., Ntombela, T., Jhamba, L. A., Maguire, G. E. M., Govender, T., Naicker, T., & Kruger, H. G. (2021). Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. European journal of medicinal chemistry, 224, 113705. https://doi.org/10.1016/j.ejmech.2021.113705
[4]Pang, M., He, W., Lu, X., She, Y., Xie, L., Kong, R., & Chang, S. (2023). CoDock-Ligand: combined template-based docking and CNN-based scoring in ligand binding prediction. BMC bioinformatics, 24(1), 444. https://doi.org/10.1186/s12859-023-05571-y

















 1600
1600

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









