










原创 普美瑞生物科技 普美瑞生物 2024年11月27日 09:45 江苏
背景介绍
动脉粥样硬化(Atherosclerosis)是一种常见的血管炎症性病变,是多种心血管疾病的根本病理基础,如高血压、心肌梗死、中风和外周动脉疾病。已有研究表明,血管平滑肌细胞(VSMC)的功能异常与动脉粥样硬化的发生和进展密切相关,而凝血酶(thrombin)通过促进VSMC的迁移和增殖,在动脉粥样硬化中发挥着重要作用。丝氨酸蛋白酶抑制剂(Serpins)是血浆中一个复杂且广泛分布的蛋白超家族,其中一些Serpins通过抑制凝血酶,参与血液凝固、纤维蛋白溶解和炎症等关键生理过程。然而,Serpins在动脉粥样硬化中的具体作用尚未得到充分阐明。
研究简述
东南大学医学院附属中大医院心内科团队以鼠源Serpina3c和凝血酶(thrombin)为研究对象,采用实验研究与蛋白-蛋白对接技术相结合,探讨了Serpina3c与凝血酶的相互作用机制,并深入分析了Serpina3c在动脉粥样硬化中的作用。本研究对于疾病治疗和临床医学领域具有重要意义。
研究人员利用小鼠模型,通过基因敲除或过表达Serpina3c,观察其在动脉粥样硬化中的作用,结果表明Serpina3c的表达水平与动脉粥样硬化的严重程度呈负相关。Serpina3c显著减少了动脉粥样硬化斑块的形成,并减轻了血管内皮损伤。此外,Serpina3c的凝血酶抑制作用有效降低了血栓形成,缓解了与凝血相关的炎症反应。
计算与结果分析
为了进一步探讨Serpina3c与凝血酶(thrombin)之间的相互作用机制,研究人员利用普美瑞云平台(https://pumeirui.com/)上的蛋白-蛋白对接工具,预测了Serpina3c与凝血酶的复合物结构,并详细解析了结合界面上氨基酸残基的分布特征。
操作简述:
准备受体蛋白和配体蛋白文件:从晶体结构(PDB ID: 2PV9)提取thrombin的3D结构作为受体蛋白,使用MODELLER构建小鼠Serpina3c的结构作为配体蛋白。
设置可选项:采用全局对接方式,设置配体蛋白旋转角度为15°,使用基于知识的评分函数进行打分,输出打分前10的结合构象并进行优化。
下图展示了Serpina3c与凝血酶(thrombin)相互作用的复合物结构(绿色表示Serpina3c,橘色表示thrombin)。复合物的结合模式显示,Serpina3c的Loop区插入至凝血酶的活性口袋中,类似于其同系物抗凝血酶-III(antithrombin-III)与凝血酶的晶体复合物结构。此外,研究还发现Serpina3c上的多个关键氨基酸残基与凝血酶活性位点相互作用,从而维持了Serpina3c与凝血酶之间的紧密结合。
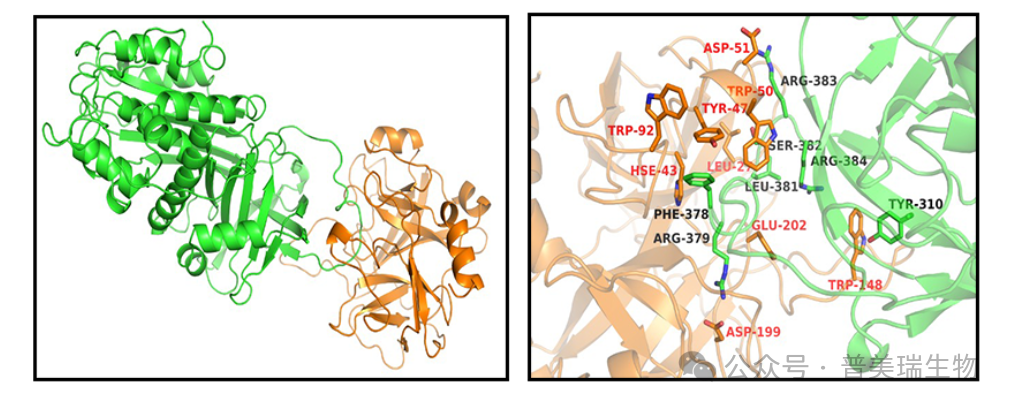
从上述研究结果,我们可以看到蛋白-蛋白对接方法展现出了强大的预测能力,能够精确地模拟出配体蛋白(Serpina3c)与受体蛋白(thrombin)之间的结合模式。这种方法不仅详尽地解析了结合界面的残基分布特征,还通过高精度的计算分析,鉴别出了对复合物稳定性及结合亲和力具有关键贡献的热点残基。这些热点残基在Serpina3c与thrombin的相互作用中扮演着至关重要的角色,它们的存在和分布直接影响了复合物的形成、稳定性和功能。通过深入解析这些热点残基的结构和功能特性,我们可以更深入地理解Serpina3c如何有效地抑制thrombin的活性,并揭示其在调节凝血和抗炎过程中的分子机制。
案例总结
本研究结合实验与计算方法,深入探讨了Serpina3c与凝血酶(thrombin)之间的作用机制。结果表明,Serpina3c通过靶向并抑制凝血酶,进而减少血管平滑肌细胞(VSMCs)的增殖和迁移,在动脉粥样硬化中发挥重要作用。该研究成果已发表于学术期刊《Clinical Science》。
参考文献
Qian L, Ji J, Guo J, et al. Protective role of serpina3c as a novel thrombin inhibitor against atherosclerosis in mice[J]. Clinical Science, 2021, 135(3): 447-463.

















 1142
1142

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









