









1.FastQC的作用
- 在建库过程或者在测序仪测序中存在的数据问题或者数据偏移问题,从而得到QC报告
- drop down selector
- FastQC官方教程
- 非root用户 Linux上安装FastQC
参考网页1
参考网页2
2.FastQC结果解读
tips:了解你的数据是一次数据分析的开始,也是贯穿整个分析流程的重要环节

颜色解读
(1)绿色——PASS
(2)黄色——WARN
(3)红色——FAIL
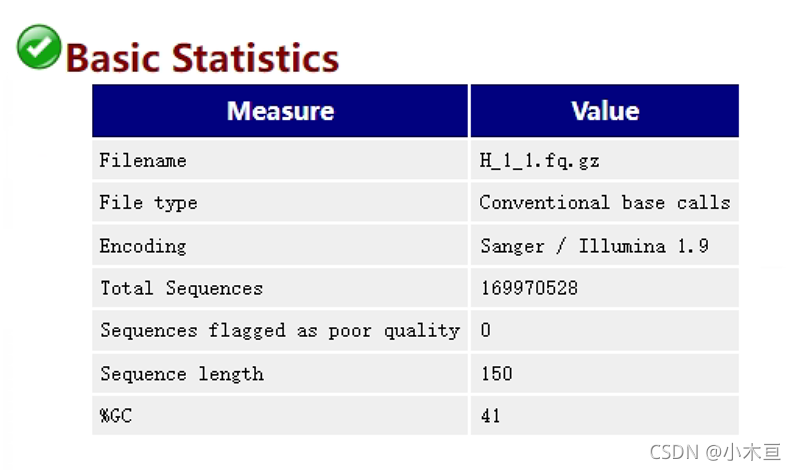
Basic Statistics——对数据的概览
1. 图表解读
- 1. Filename:文件名——H_1_1.fq.gz(这里文件名表述H类的1号样本的reads1端测序)
- illumina双端测序:一个样本包括从reads1端和reads2端两端开始测序
-Pair End测序优势:
除了序列本身外还有中间的距离信息。距离信息可以用来判定组装后
成对reads间的序列是否准确,也可用来帮助组装。
[PE&SE问题](https://www.jianshu.com/p/5c238ea7c52f)
- 2. File type:文件类型——常规碱基识别
- 3. Encoding:测序平台——Sanger/Illumina 1.9
- 这里的测序平台可以关注一下,主要是与碱基质量体系标准相关。
- 因为早期碱基质量体系没有统一的标准,有的加33,有的加64,
不同测序仪可能不同,所以有Phred33和Phred64两个质量体系,
现在基本上统一为Phred33体系了。
- 4. Total Sequences:reads总数
- reads是高通量测序平台产生的序列标签,翻译为读段
- 5. Sequences flagged as poor quality:标记为低质量的序列
- poor quality 只是针对Casava格式的序列,
- 对于illumina平台的fastq格式的数据,没有这一项统计内容,值永远为0。
- 6. Sequence length:测序长度——150bp
- 一般会给最大长度和最小长度,如果一样长,则只给一个值
- illumina会限制合成链的长度,测序长度一般为150bp(<Sanger的1000bp):
- 1. 由于测序过程中存在碱基未配对或错配,从而在荧光信号中
出现杂信号的情况,且测序长度越长,杂信号越多,当杂信号过多,
系统无法识别时,只能给N,而N过多对于后续的数据处理很麻烦,
去除会丢失数据,不去除会造成冗余。
- 2. 随着测序的不断推进,酶活性逐渐降低,则序列的错误率也不断增加。
- 7. %GC[重要]:GC含量——可以帮助区别物种(其中人类细胞GC含量大致为42%左右)
Per base sequence quality——该样品reads中每个碱基测序的质量值[非常重要]
-
基本概念
(1)碱基质量值(Quality Score或Q-score):-
定义:碱基识别(Base Calling)出错的概率的整数映射。
-
通常使用的Phred碱基质量值公式为:
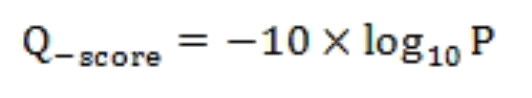
其中,P为碱基识别出错的概率
-
碱基质量值与碱基识别出错的概率的对应关系表:
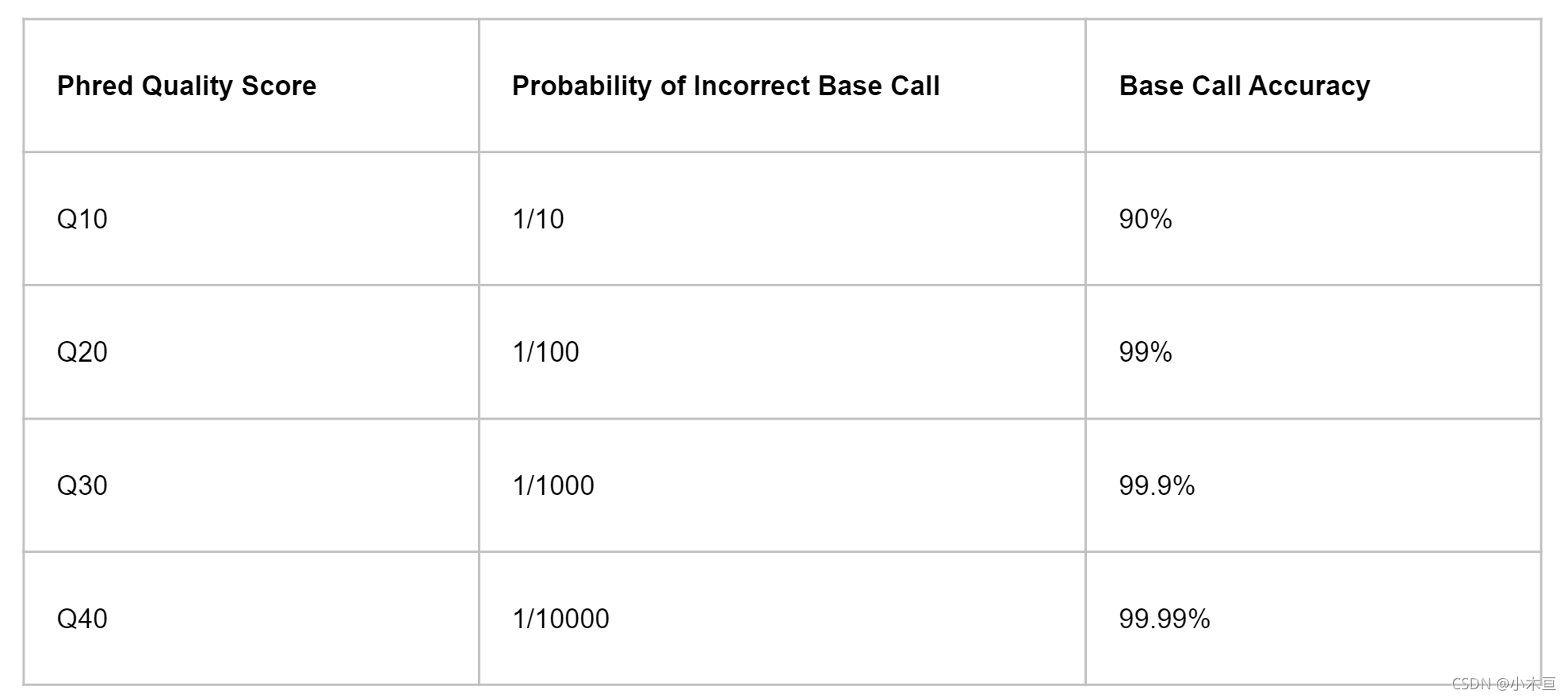
对应表解读:- 碱基质量值越高表明碱基识别越可靠,碱基测错的可能性越小。 - 1)**Q20**的碱基识别,100个碱基中有1个会识别出错; 2)**Q30**的碱基识别,1,000个碱基中有1个会识别出错; 3)**Q40**表示10,000个碱基中才有1个会识别出错。(2)FastQC中read碱基质量值分析:
- FastQC并不单独查看具体某一条read中碱基的质量值,而是将一个Fastq文件中所有的read 数据都综合起来一起分析。
(3)碱基质量值过低时的处理方法:
- 最好的情况是重新测序
- 但如果不得不使用这个数据,就要把这些低质量的数据全都去除掉才行
- 同时还需留意是否还存在其他的问题,但不管如何都一定会丢掉很大一部分的数据。
-
-
图表解读
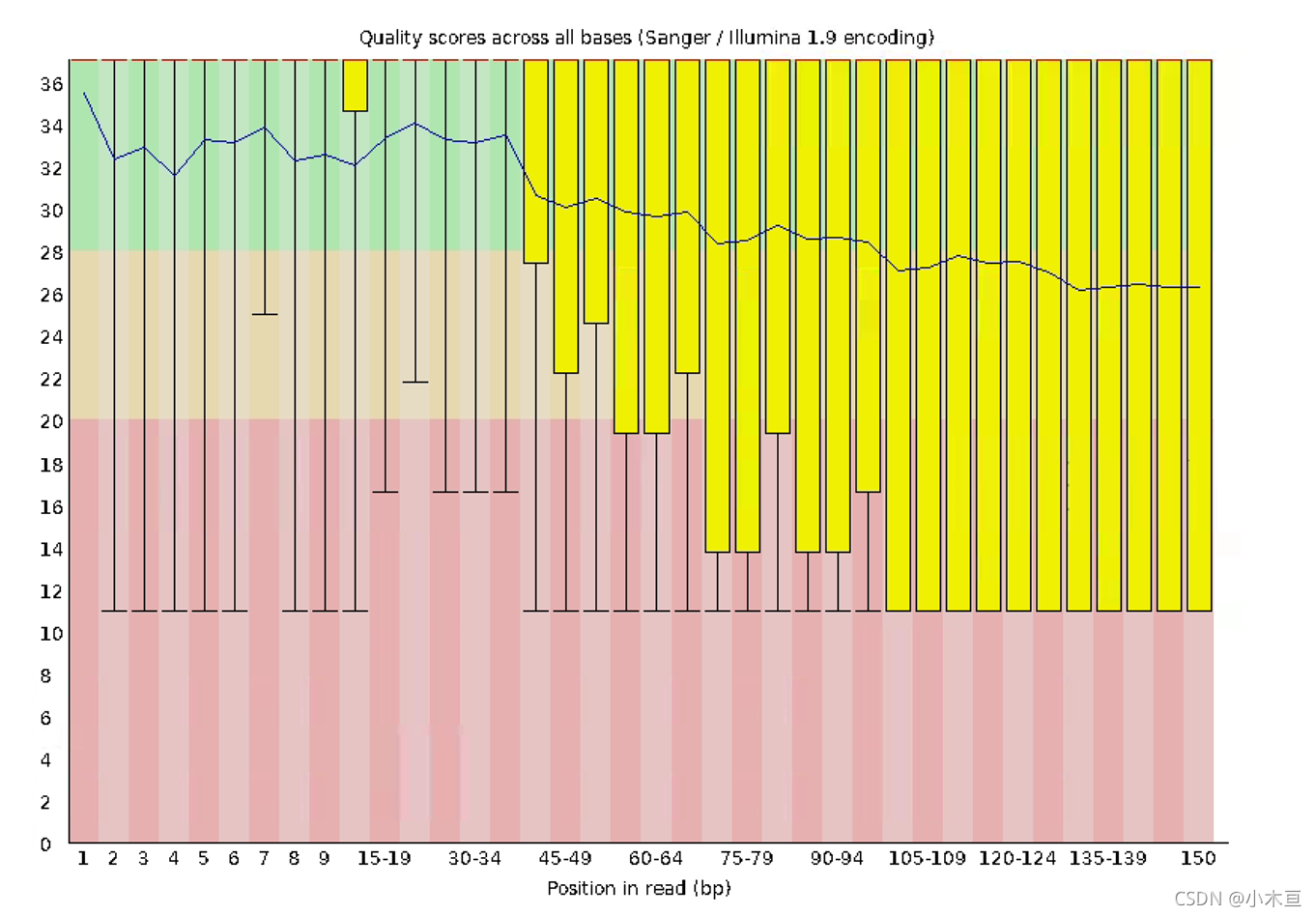
- 1. 背景色: - 0-20:背景色为红色,测序质量非常糟糕 - 20-28:背景色为橘色,测序质量差 - 28以上:背景色为红色,测序质量良好 - 2. 横纵轴 - 横轴:测序序列从1到第150个碱基 - 纵轴:质量得分,Q = -10*log10(error P,即20表示1%的错误率,30表示0.1% - 2. 箱式图 - 每1个boxplot:该位置的所有序列的测序质量的统计 (tips:FastQC并不单独查看具体某一条read中碱基的质量值, 而是将一个Fastq文件中所有的read数据都综合起来一起分析) - 上面的up bar——90%分位数,下面的down bar——10%分位数 - 箱子中间的横线——50%分位数,箱子顶upside——75%分位数, 箱子底downside——25%分位数 - boxplot的意义: - 1. 看数据是否具有对称性; - 2. 看每个碱基位置数据分布差异(这里主要利用了第二点); - bar的跨度越大,说明数据越不稳定 - 3. 蓝色细线——各个碱基位置平均值连线 - 图中蓝线的走势解析? - 因为目前采用的边合成边测序使用的是化学方法促使链由5'向3'延伸, 也就是利用了DNA聚合酶。刚开始测序,合成反应还不是很稳定, 但是酶的质量还很好,所以会在高质量区域内有一定波动(这里的1-30bp) 后来稳定了,但是随着时间的推移,酶的活力逐渐下降,特异性也变差, 所以越往后出错几率越大。 - 4. Q20过滤:所有位置的10%分位数大于20 - 不满足Q20过滤的序列需要被切掉,从而保证后续分析的正确性。 - 二代测序,最好是达到Q20的碱基要在95%以上(最差不低于90%); Q30要求大于85%(最差也不要低于80%) - 5. 得到的结果报警状态: - Warning 报警:如果任何碱基质量低于10,或者是任何中位数低于25 - Failure 报错:如果任何碱基质量低于5,或者是任何中位数低于20 - Pass:其他 - 6. 通常,在序列的起始和结束部分可能出现质量较差的情况 - 7. 碱基质量值在开头存在上升趋势,随后下降: - 在序列的起始和结束部分可能出现质量较差的情况,对于最初测序的部分数据, 测序仪直接使用默认参数进行base calling, 这部分碱基的质量一般, 然后会 利用这部分数据去调整base calling的参数设置,以符合真实的数据,在之后的 测序中,用调整后的参数进行base caling, 此时碱基的质量会更好,所以观察 到,在开头部分存在碱基质量上升的趋势;随着测序反应的进行,酶活性等 因素降低,会导致测序质量变差,所以在结尾部分会观察到碱基质量降低趋势。
Per tile sequence quality——每个荧光信号识别小孔测序质量统计

- 1. 横纵轴
- 横轴:1-150个碱基的位置
- 纵轴:tail的index编号
- 2. 面板中的条纹颜色
- 蓝色—代表测序质量高
- 暖色—代表测序质量不高
当某些tail出现暖色,后续分析中把该tail的测序结果全部去除
- 3. 此图的意义:
为了防止在测序过程中,某些tail受到不可控因素影响而出现测序质量偏低
Per sequence quality scores——每条序列的测序质量统计
每条reads的测序质量:该reads上所有碱基质量平均值
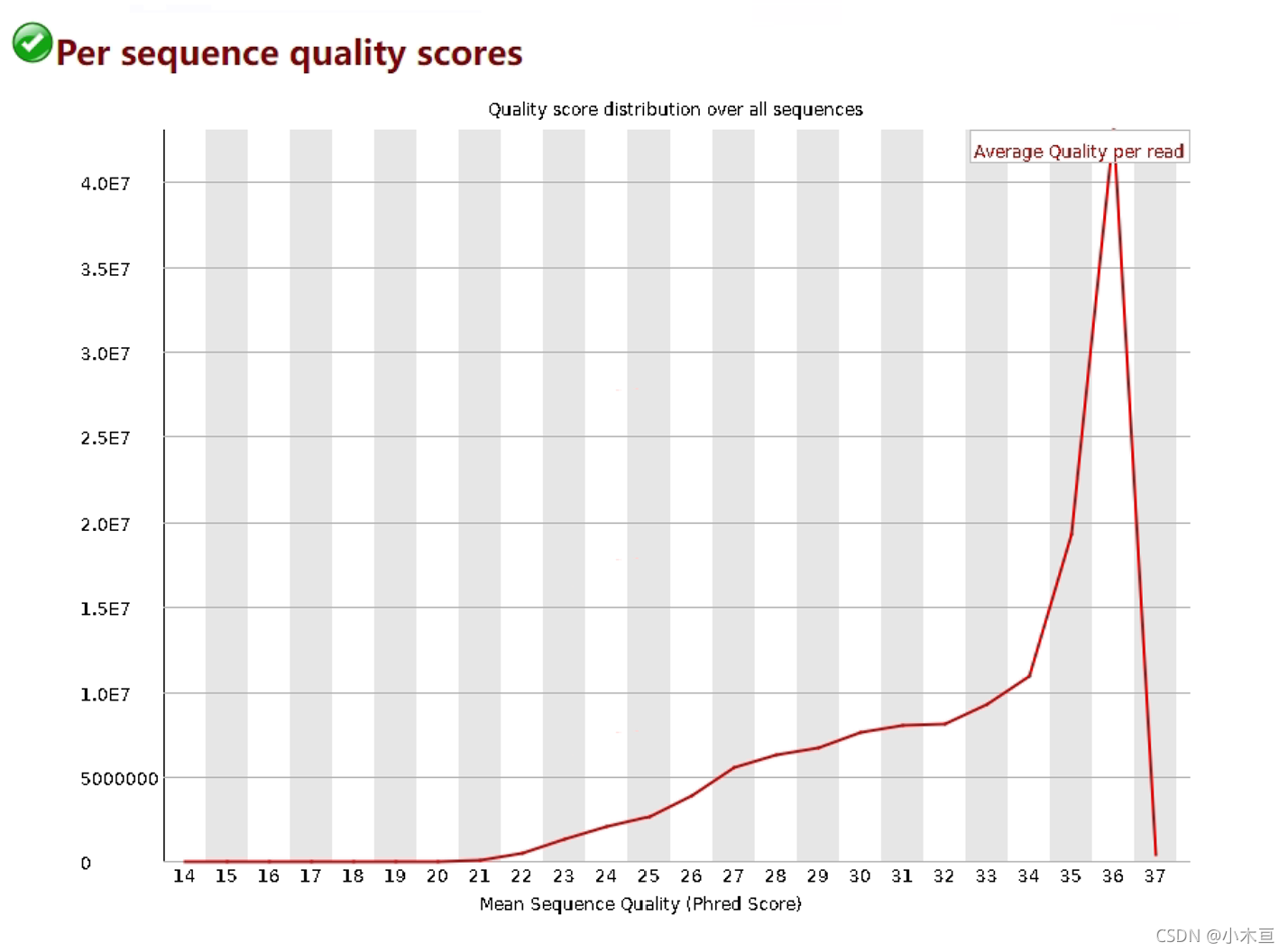
- 1. 横纵轴:
- 横轴:Q值
- 纵轴:每个Q值对应的reads数
- 2. 结果判读:
- 只要大部分高于20,说明比较正常
- 如果测序结果集中在高分段,则说明测序结果良好
- 一般认为90%的reads测序质量在35分以上,则认为该测序质量非常好
- 报"WARN":当测序质量峰值小于27(错误率0.2%)
- 报"FAIL":当峰值小于20(错误率1%)
Per base sequence content——reads每个位置上ATCG的比例分布
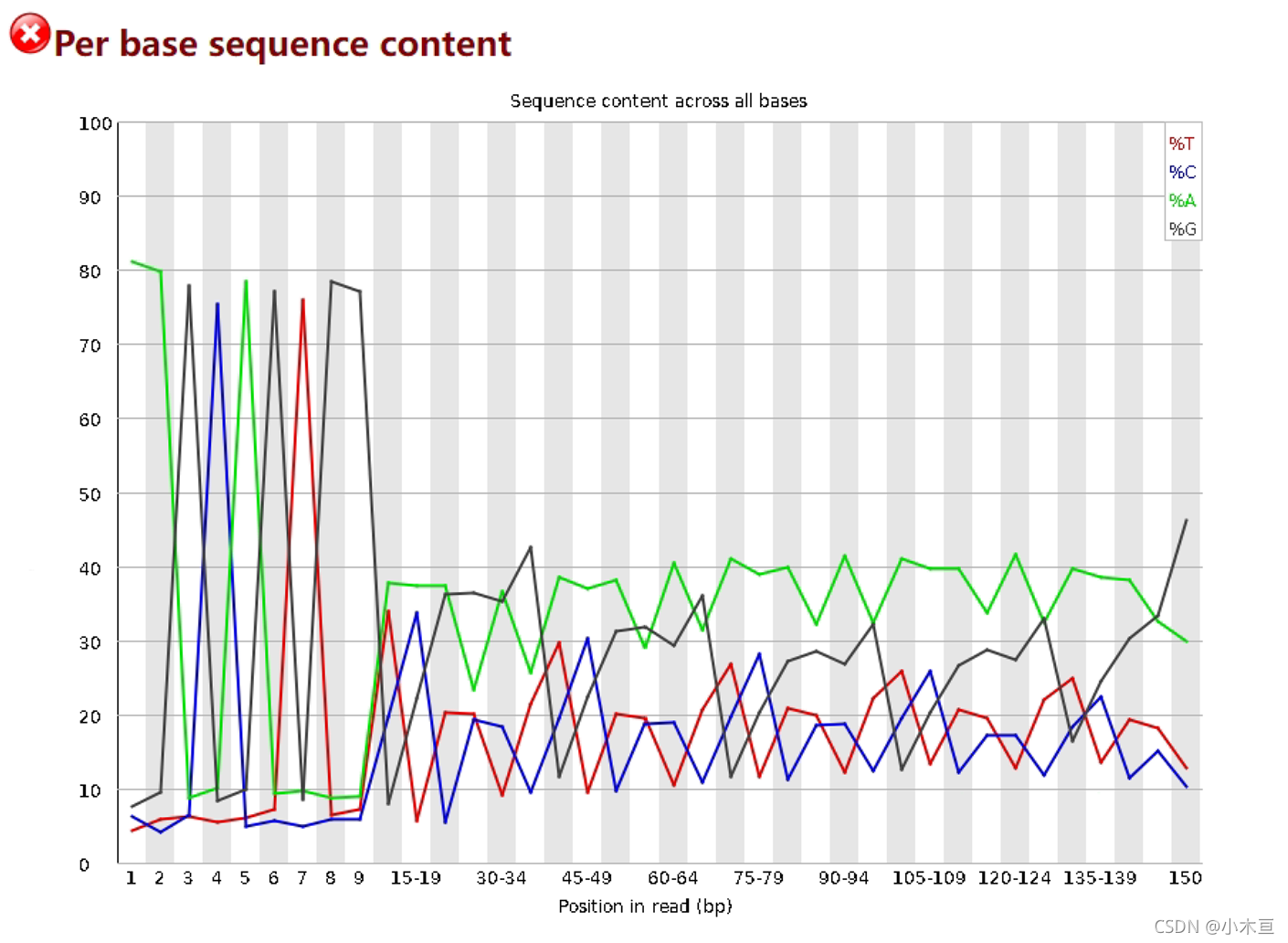
- 1. 横纵轴:
- 横轴:各碱基位置
- 纵轴:碱基百分比
- 2. 四个颜色线条:每种碱基在每个位置的平均含量
- 3. 一般来讲,A=T, C=G, 但是刚开始测序仪不稳定可能出现波动,这是正常的。
一般不是波动特别大的,像这里cut掉前5bp就够了。另外如果A、T 或 C、G间出现偏差,
只要在1%以内都是可以接受的。
- 4. 理想情况下,各个碱基的比例并不会随着测序反应的进行发生变化,所以每个碱基对应的线
应该是相互平行的,而且对于碱基随机分布的文库,A和T碱基数量相等,G和C碱基数量相等。
- 5. Warn:当A和T或者G和C的比例相差超过10%时,会给出警告信息,
Fail:当A和T或者G和C的比例相差超过20%时,会给出错误信息。
- 6. 碱基分布偏倚可能存在的问题:
- 当文库中引物二聚体序列比例很多时,这种情况就是文库的构建过程存在问题了。
- 当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有
overrepresented sequence的污染。
- 当所有位置的碱基比例一致的表现出bias时,即四条线平行但分开,往往代表文库有
bias (建库过程或本身特点),或者是测序中的系统误差。
- 对于亚硫酸氢盐处理的甲基化测序文库,未甲基化的C会转换成T,也会出现碱基分布的偏倚。
Per sequence GC content——序列的GC含量分布
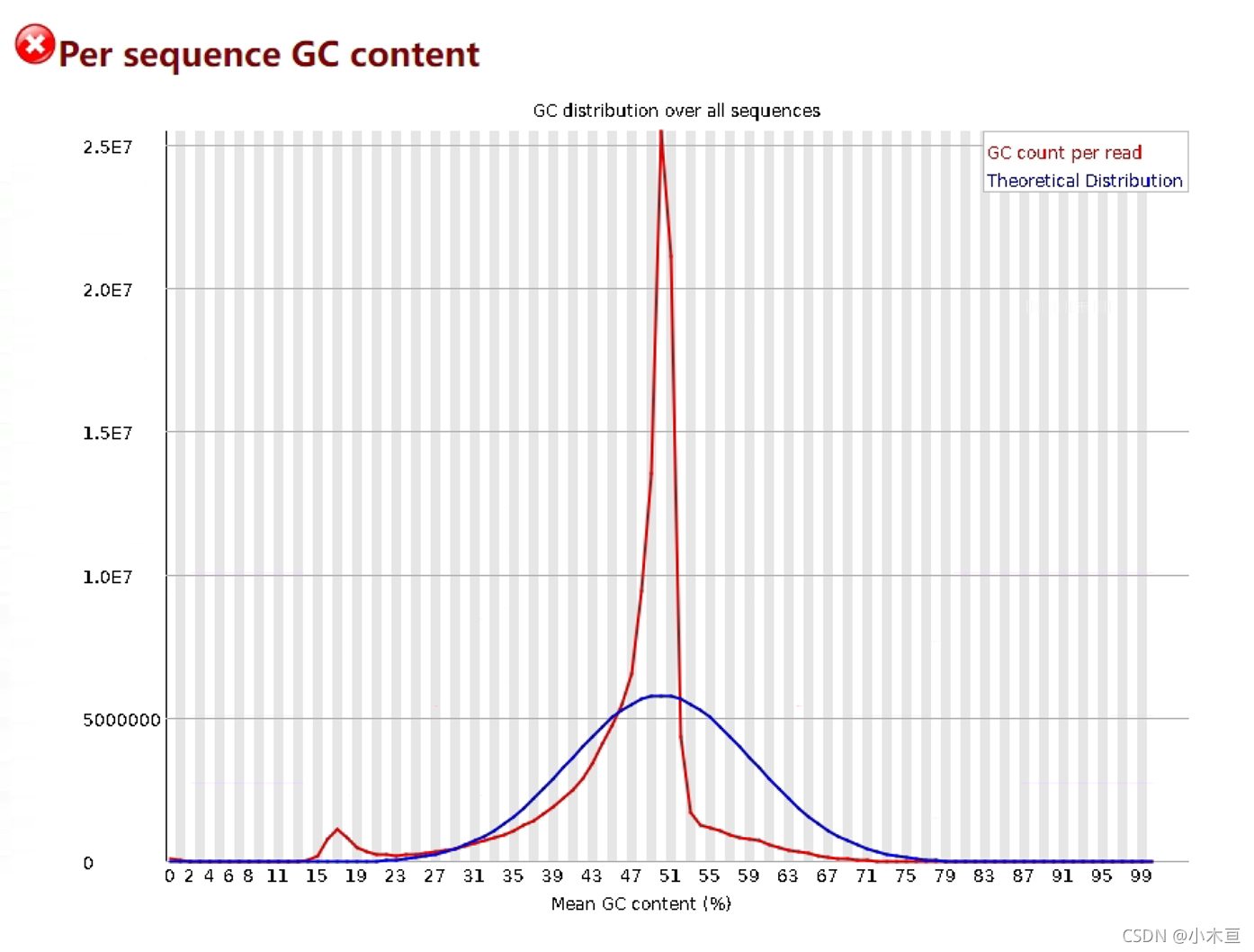
- 1. 横纵轴
- 横轴:平均GC含量
- 纵轴:每个GC含量值对应的reads数
- 2. 线条
- 蓝色:为系统得到的理论分布情况—为正态分布
- 红色:为实际分布情况
- 这两条线越接近越好
- 3. 报错
- Warn:如果实际曲线偏离理论分布的总和大于15%
- Fail:当实际分布曲线偏离理论分布曲线的总和大于30%
- 4. 仅出现正态分布的偏移时,提示是与碱基位置无关的systematic bias,
这不能说明是错误,这种偏移与所测基因组的物种有关(物种不同,GC含量不同)
- 5. 报错原因
- 1. GC可以作为物种特异性根据
- 当红色的线出现双峰或多峰
- 平滑曲线上比较宽阔的峰——混入了其他物种的DNA序列
- 平滑曲线上出现尖峰时——提示明确的污染物混入
- 如adapter二聚体(这在overrepresented sequence会被提及)
- 2. other kinds of biased subset
- 目前二代测序基本都会有序列偏向性(所说的 bias),也就是某些特定区域会被反复测序
- 以至于高于正常水平,变相说明测序过程不够随机。
- 部分reads构成的子集有偏差(overrepresented reads)
- 这种现象会对以后的变异检测以及CNV分析造成影响
Per base N content——N含量分布
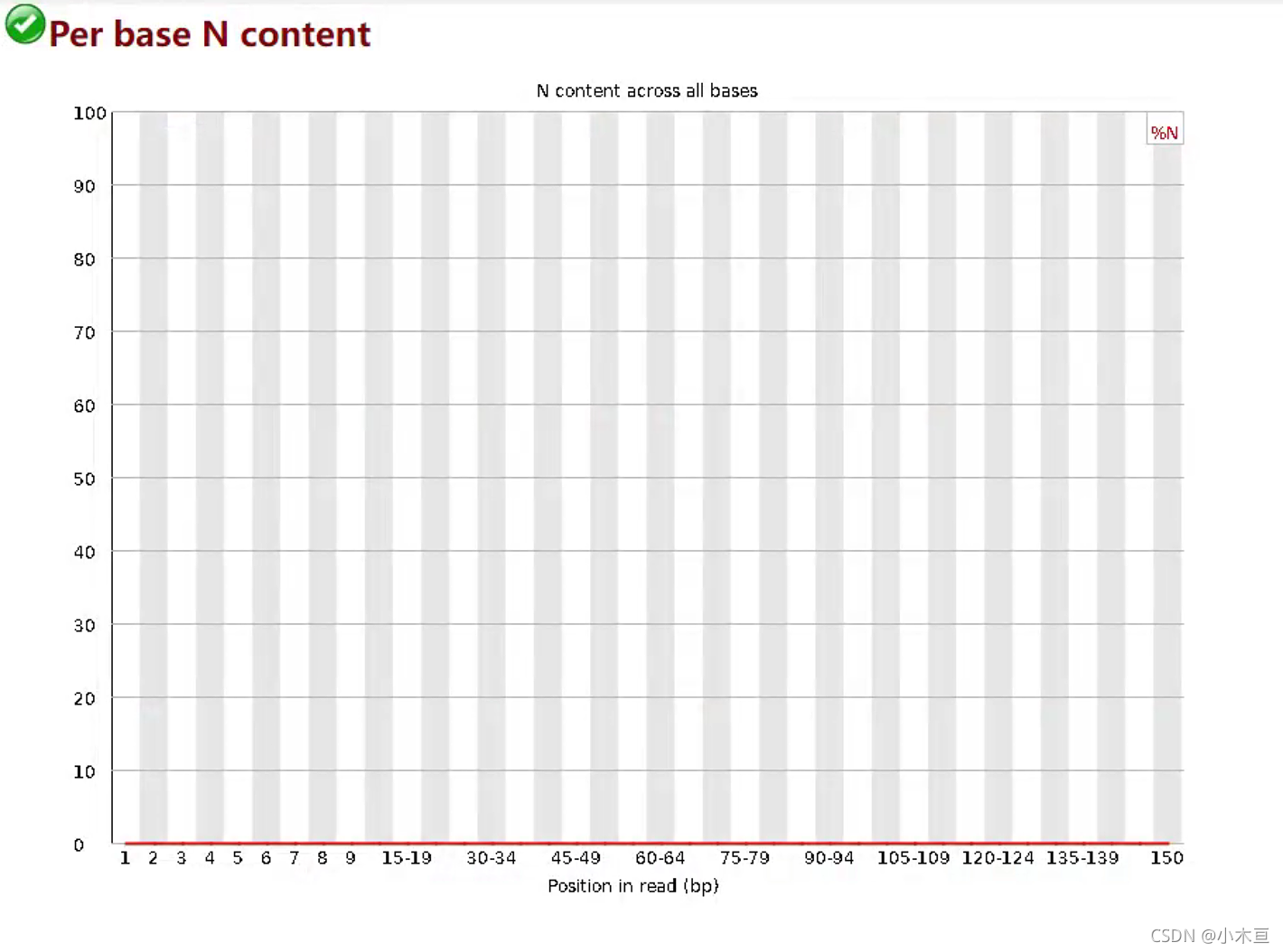
- 1. N:是在测序仪无法识别ATCG碱基时,给出的值
- 2. 报错:
- WARN:任意位置的N的比例超过5%
- FAIL:任意位置的N的比例超过20%
- 3. 错误原因:
- 如果出现并且量还很大,应该就是测序系统或者试剂的问题
Sequence Length Distribution——测序长度统计
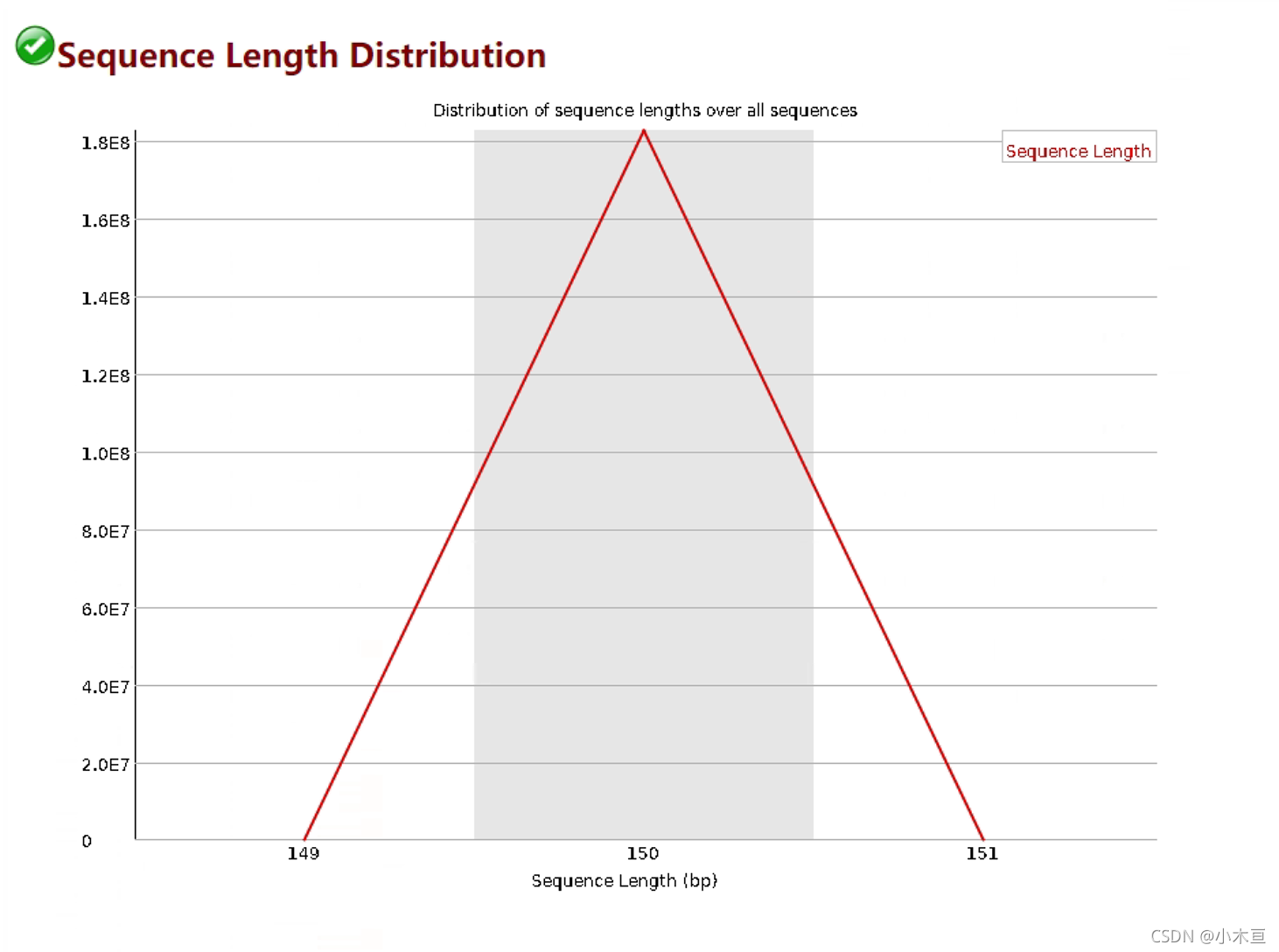
- 1. 理想情况下,测得的序列长度应该是相等的。实际上总有些偏差
- 2. 报错:
- Warn:reads长度不一致很严重时,这表明测序仪在此次测序过程中产生的数据不可信
- Fail:存在reads长度为0时
- 3. 报错原因
- 为了防止建库或者测序时有一些不规则长度的序列被进行测序
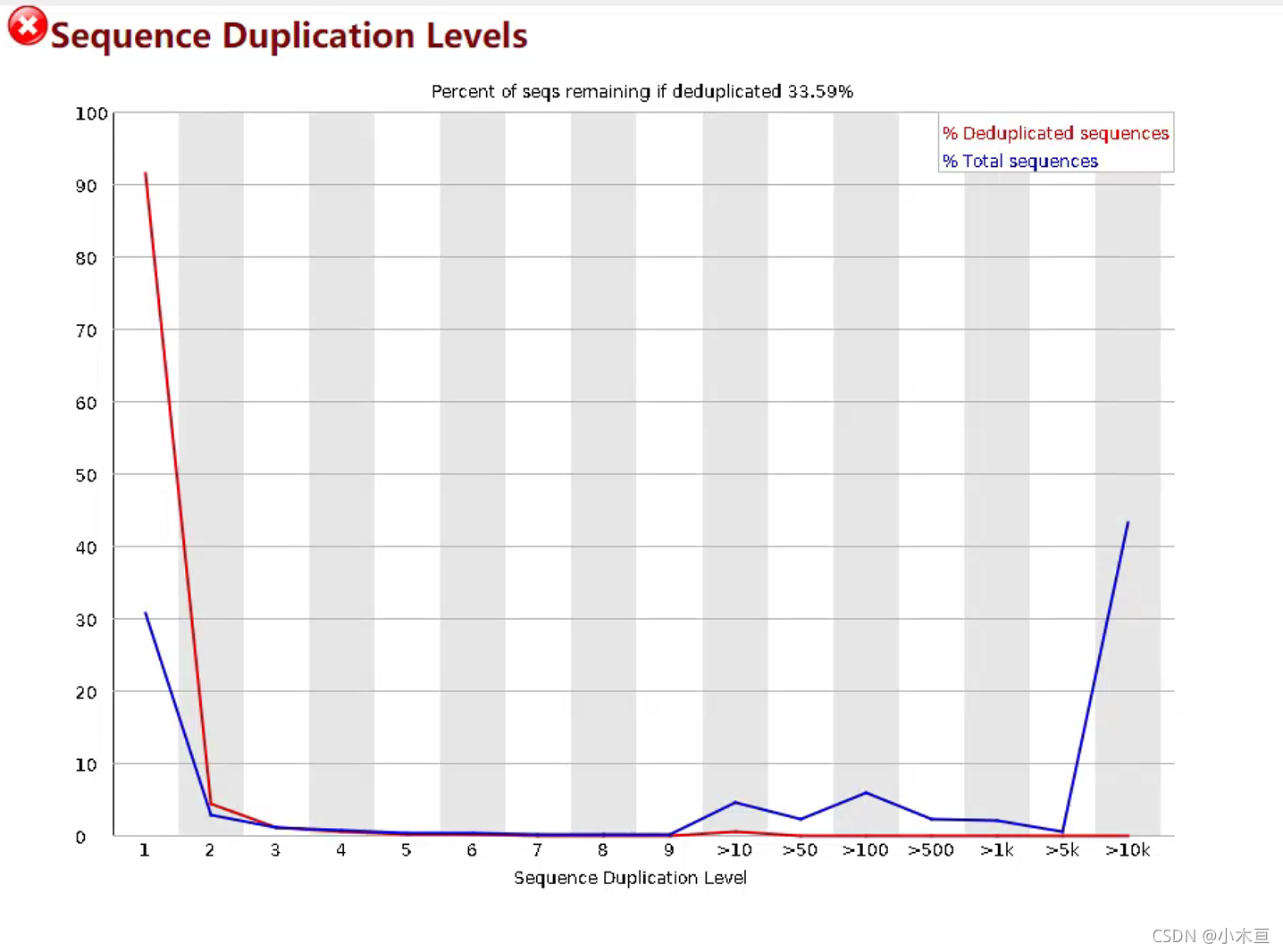
Sequence Duplication Levels——统计序列完全一样的reads频次
- 1. 横纵轴
- 横轴:duplication的次数
- 横轴上的>10,>50,>... :测序的原始数据很大,如果每一条reads都统计,将耗时很久。
这里软件只采用了数据的前100,000条reads统计其在全部数据中的重复数目,重复数大于10
的reads被合并统计成了>10,以此类推...
- 另外大于75bp的reads只取50bp进行比较(因为对于复制序列的判断需要序列全长完全匹配,
所以对于长度大于75bp的序列只取前50bp)。重复数大于10的reads被合并成了>10,以此类推...
- 纵轴:duplicate rate—duplicated序列占全部序列百分比
- duplication rate = 1 - unique reads / total reads
- 2. 图标
- 红线:deduplicate【去duplicate之后序列理论重复性分布(服从泊松分布或者二项分布)】
- 就是看,去除了建库过程中产生的duplicate(PCR duplicate,cluster duplicate,
光学duplicate,正负链duplicate)后,剩下的reads中存在duplicate的情况(这应该就是
生物学上的duplicate)
- duplicate reads定义:
- reads的起始和终止位置一样,起点和终点之间的碱基序列一样(不妨简称为“三一样”)
- duplicated reads是PCR对同一个分子进行多次镜像复制的后果
- 蓝线:unique reads总数
- unique reads
- 只要起点、终点、或者起点与终点之间的序列中有一个不同,就是不同的分子,为unique reads
- 3. 报错
- Warn:非unique的reads占总数的比例大于20%
- Fail:非unique的reads占总数的比例大于50%时
- 4. 原因分析
- 正常情况下,测序深度越高,越容易产生一定程度的duplication。
- 但当duplication level的程度过高时,提示我们可能有bias的存在
- (如建库过程中的PCR duplication)。
- 为什么文库构建过程中 PCR 将每个文库分子都扩增了上千倍(以 PCR 10个循环为例 2^10= 1024)
但是实际测序数据中 duplication 率并不高(低于20%)?
- 文库中 unique 分子的数量比被 flowcell 上引物捕获的分子数量多很多,
直白点说就是 flowcell 上用于捕获文库分子的引物数量太少了,两者不在同一个数量级,
导致很少出现同一个文库分子的多个拷贝被 flowcell 上引物捕获生成 cluster。
- 反转录文库大部分序列一般只出现一次。
- 低水平的复制次数也就代表着对于目标序列高水平的覆盖度;
高水平的复制次数更像是富集偏倚(如PCR扩增过度)
- 一般转录组测序的结果中duplication level都比较高,60-70%都正常,这是因为转录组测的是
基因的覆盖深度,各个基因表达量不同,如果某个基因覆盖度较高【tip:覆盖度是指基因/转录组
测序测到的部分占整个组的比例】,那么测的部分就越多,相对应的duplication也会更高;
而对于外显子组测序来说,一般覆盖度比较一致,这里出现了duplication就不太正常。
- 基因组覆盖度越高,测序得到的序列重复比例会越低;
- 在文库构建过程中,如果某些片段PCR扩增的比例大于随机扩增的比例,会导致重复序列比例高。
- reads越长越不容易完全相同(由测序错误导致),所以其重复程度仍有可能被低估。
- remove duplicate的情况
- RNA-seq中,一般不考虑deduplication(有paper专门讨论过这个问题)
- 在RNA-seq数据中,可能存在某些转录本较短,表达水平很高,导致随机打断后被重复抽到测序
的概率大大增加。因此对于RNA-seq数据来说,重复性较高的序列不一定就是PCR重复。
- 低水平的重复:目标序列的覆盖度较高
- 高水平的重复:PCR过度扩增—出现了富集bias
- DNA-seq中,序列如果是随机打断,需要考虑deduplication;酶切样本一般不考虑这个问题
- 蓝线右侧出现尖峰,而红线没有时
- 特异富集的子集,或低复杂性污染物的存在,将倾向于在右侧产生尖峰。
- 如果要对文库进行重复数据删除,该模块还会计算预期的总体序列丢失。
- 这一标题数字显示在图的顶部,给人一个合理的印象,可以看出潜在的整体损失水平
- 实验问题
- PCR扩增次数太多或者起始扩增底物太少,都会降低文库的复杂度
- 5. 针对本图分析
- 1. 本图为RNA-seq文库
- 2. 表头可知:在去除重复之后,剩下的序列为total sequences的33.59%
- 3. 观察两条折线可知:
- 在重复次数为1时,deduplicated sequences达90%以上,而total sequences仅30%左右,
说明此测序中duplicate的程度很高 ;在>10重复次数的序列中,deduplicated sequences
比例逐渐趋于0,而total sequences 所占比例却在5%左右,尤其是>10k的序列比例,
达到了40%,说明有极少数序列重复了上万次,导致整个文库的重复率特别高,这少部分
的序列应该是PCR重复所致。
Overrepresented sequences——大量重复序列
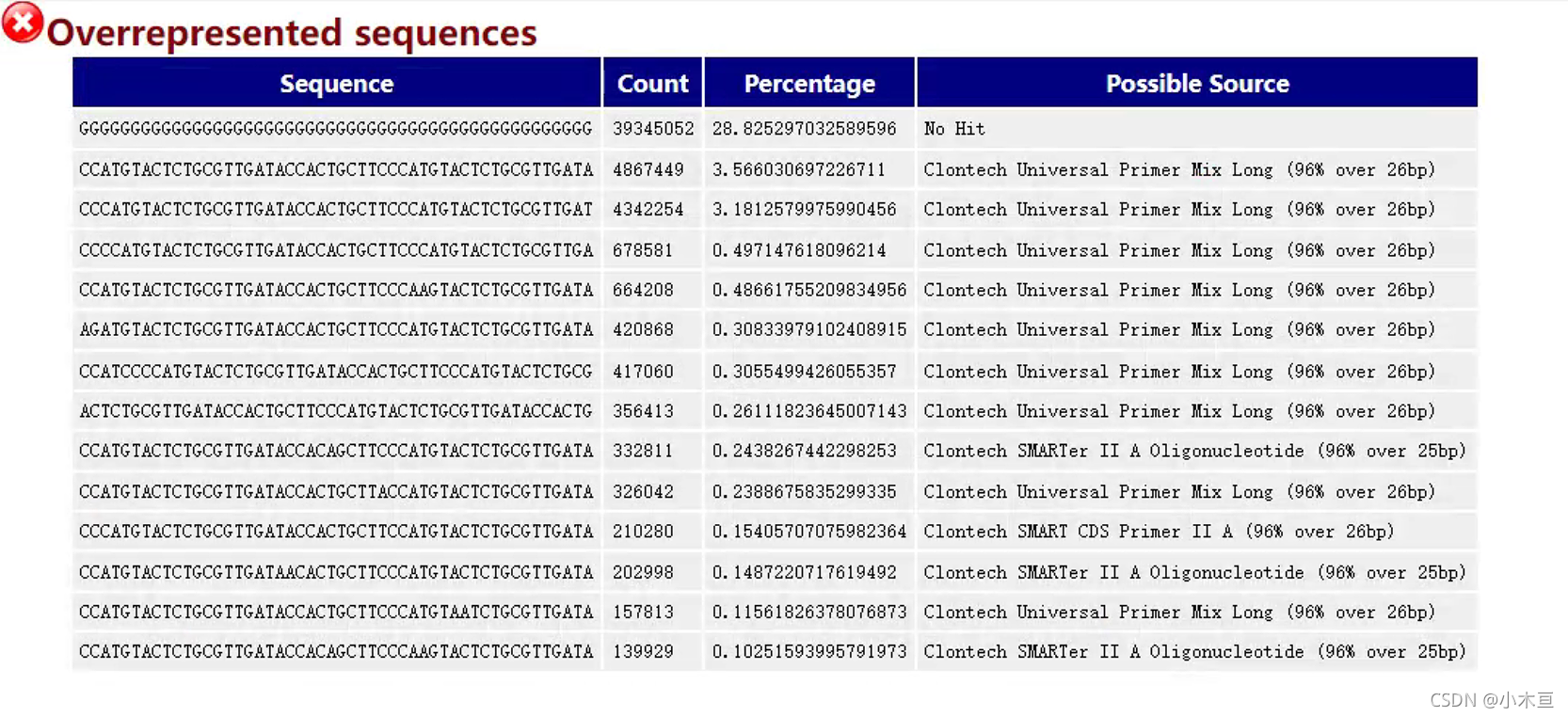
- 1. 一个正常的高通量文库将包含一组不同的序列,单个序列只占整个文库的一小部分。
- 发现单个序列在集合中的代表性非常高时:
- 1. 它具有高度的生物学意义
- 2. 表明该文库受到污染(载体,接头序列)
- 程序会自动在污染物的数据库中找到匹配项
- 3. 没有预期的那么多样化
- 2. 样本选择:
- 1. 选取的reads是那些占比超过total sequence 0.1%的
- 2. 只采用了数据的前100,000条reads统计
- 可能在100,000条reads之后的reads中,也存在过表达的情况,这种情况就会被忽视
-
-
Adapter Content
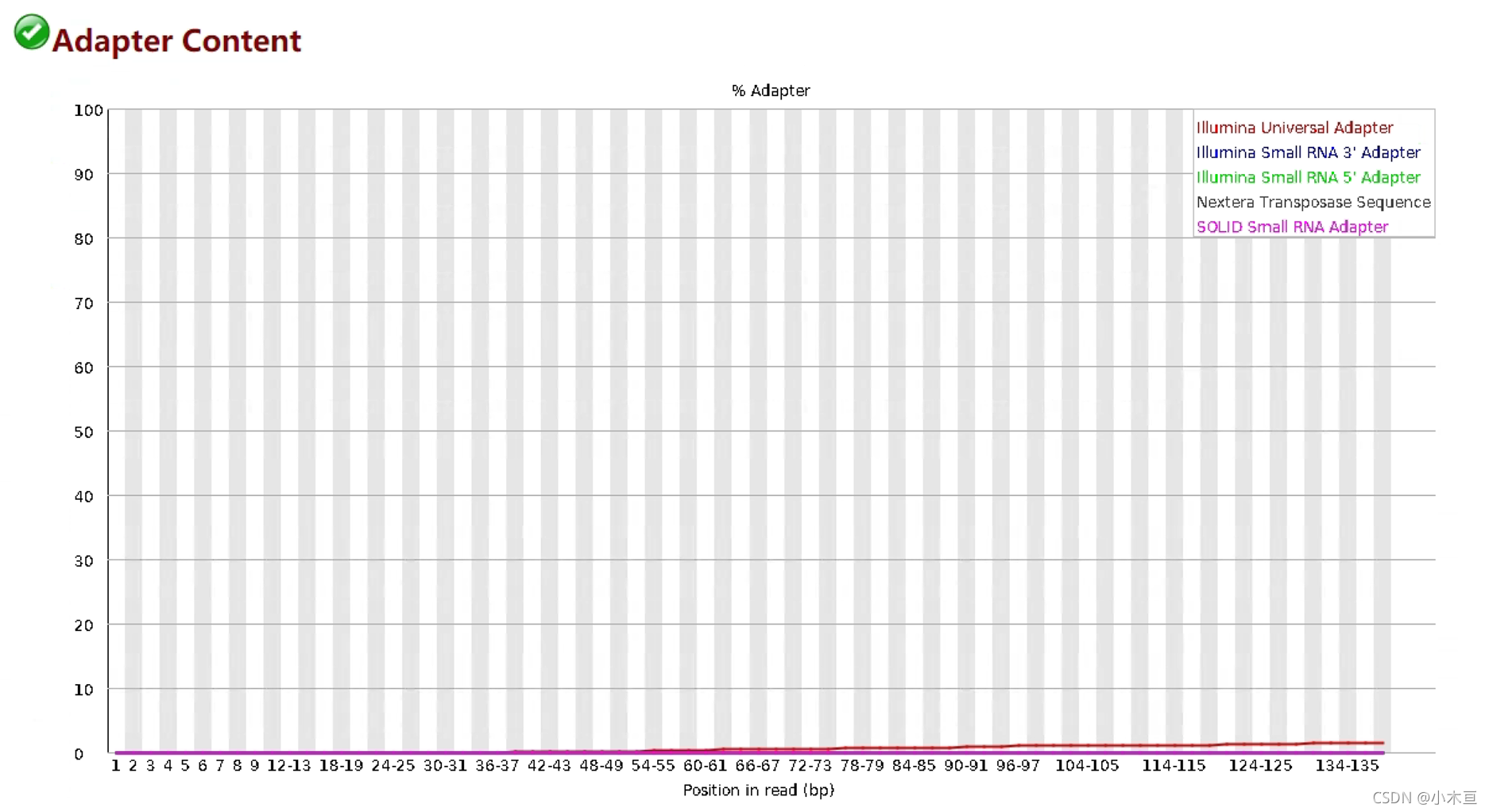
-1. 与在下游分析中,序列是否需要对接头进行剪接有关
REFERENCE:
1. 测序数据质量控制
2. 测序的世界
3. 孟浩巍—使用FastQC测序分析
4. NGS中的duplicate问题
5. FastQC_sequence duplication详解
6. NGS_FastQC_sequence_duplication_PCR过度扩增,产生bias的实验问题

















 568
568

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









