







参考我的上一篇博客https://blog.csdn.net/weixin_62528784/article/details/142106516?spm=1001.2014.3001.5502
在hic-pro处理了hic数据,进行质控解读之后,并且获取了各种下游分析所需的数据格式之后,下一步就是在识别各层次结构compartment、TAD、loop前,对hic数据进行的上游模块分析了,
具体分析并不限制于文章中所提到的模块,待之后阅读CNS文献进行拓展;
脚本参考TNBC-project+PDXHiC_supplemental+PDX-HiC_processingScripts,
1,样本质控统计:
(1)分辨率评估:
参考juicer的脚本calculate_map_resolution.sh,分辨率bin size的概念也是该文章2014提出来的;
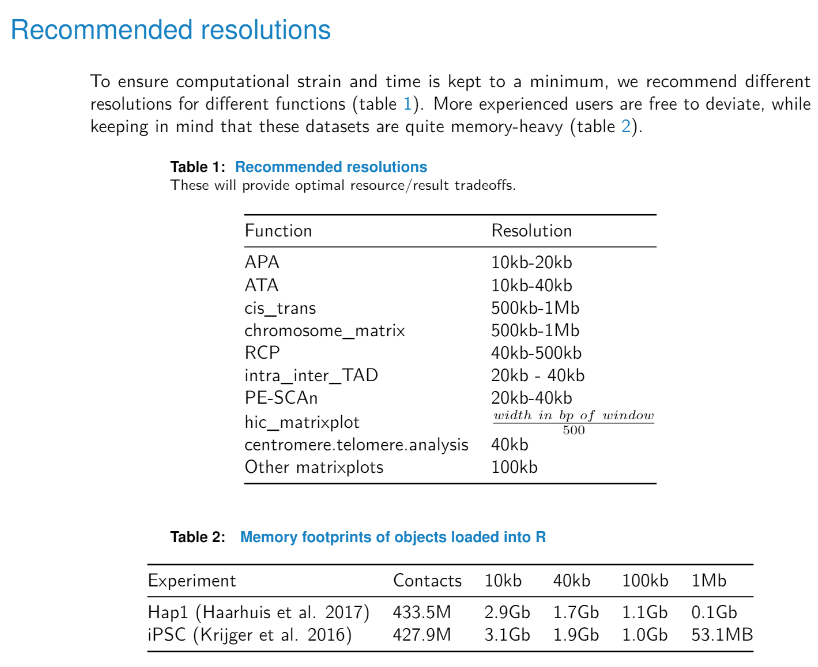
要明确的一点是,下游各个层次的数据分析以及各个子模块的分析都需要明确使用的数据分辨率是多少,即你有多少分辨率的数据,才能完成多少对应分辨率的数据分析任务;
比如说我的hic数据深度比较低、reads数据也很小,最精细的分辨率只有100kb,那你能做TAD分析吗,不能!更别谈loop了!
硬是要做也行,但是假阳性问题会比正常分析更严重!
个人一般经验:
compartment:100kb及以上;
TAD:40kb以及以下;
loop:10kb及以下。
参考https://github.com/robinweide/GENOVA/blob/master/vignettes/GENOVA_vignette.pdf
假设我们的上游数据都是hic-pro处理出来的,那如何使用改脚本处理以便于评估上游hic数据分辨率如何?
如果数据不够,一般文献中都建议合并技术性样本rep,比如说合并上游hic的fq文件,
当然hic-pro输出的数据格式allvalidpairs等也能够合并,参考对应issue或者google论坛即可。
但是一般下游合并比较麻烦,所以建议是上游直接合并,那就是cat fq;
按照提示:
https://github.com/aidenlab/juicer/issues/178
看看能不能不在使用juicer的情况下使用hic-pro中输出的valid pairs文件进行处理
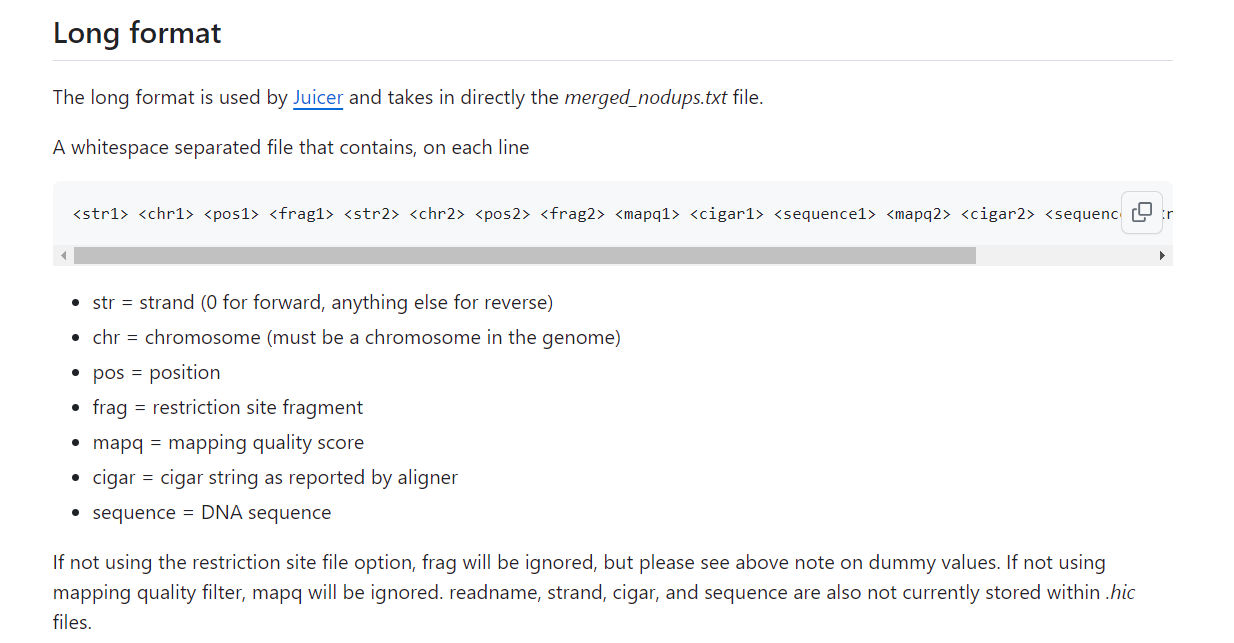
参考原始hic-pro中格式输出:
https://nservant.github.io/HiC-Pro/MANUAL.html#browsing-the-results
主要是cal所需要的原来数据格式:
https://github.com/aidenlab/juicer/wiki/Pre#file-format
如果文件转换麻烦的话,那么上游得使用juicer从头处理了,而不是使用hic-pro。
(2)hic-pro本身输出的pic文件夹中的质控结果图pdf,见上一篇博客
(3)第三方统计脚本,统计顺反互作比例:能够获取hic-pro中各步骤统计结果stat文件,并重新统计分析的,
hic_results/stats中存放的统计文件,使用脚本hicpro_collectStats.sh进行分析收集,参考<https://github.com/MaybeBio/TNBC-project/blob/main/1%2CHiC-pro%20upstream%20processing/hicpro_collectStats.sh>,
这个脚本的shell执行命令,注意执行该脚本要求hic-pro输出文件夹原封不动,
另外注意这个脚本的实际代码细节进行了修改(主要是对结果中某行的reads数目感到怀疑),原脚本参考PDX-HiC_processingScripts/5d.collectResults/中的11.hicpro_collectStats.sh+12.hicpro_mergeStats.sh,用前者11即可,然后这个脚本就是对11的某处x2修改,但是不确定是否可靠,建议使用11处理之后看统计结果reads数目正常与否,再考虑是否按照该脚本处理x2等;
统计之后的效果也就是这样: 只有mergestat有用
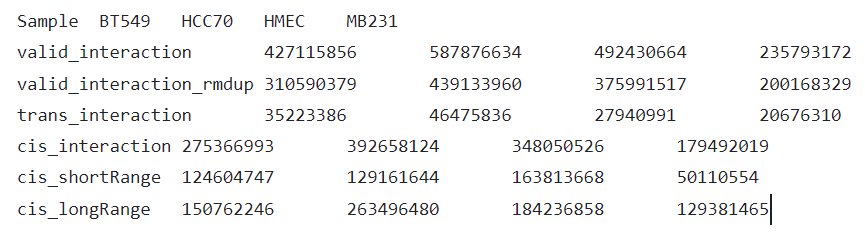
实际上就是顺反互作比例的统计;
另外也有工具可以对hic-pro结果进行统计分析:
2,hic矩阵的热图展示:
实际上hic、cool等数据格式都可以进行互作热图可视化,要么是上游的hic-pro、juicer,要么是下游的GENOVA、HITC、hicexplorer等python/R包等。
不过我建议使用juicebox,
主要是.hic文件在juicebox中进行可视化(比对)
+可以搭配后期的3层次结构track
+可以其他组学track,
实际上就是3D基因组全局差异feature比较:差异互作图谱contact map(可视化),
juicer工具使用参考https://github.com/aidenlab/juicer/wiki,然后就是软件内置readme,
juicebox工具使用,参考https://github.com/theaidenlab/juicebox/wiki,
得到.hic文件之后,如果要在win中运行juicebox,就需要下载hic文件(但是一般都是几个G,比较大);
所以有必要在linux上运行,
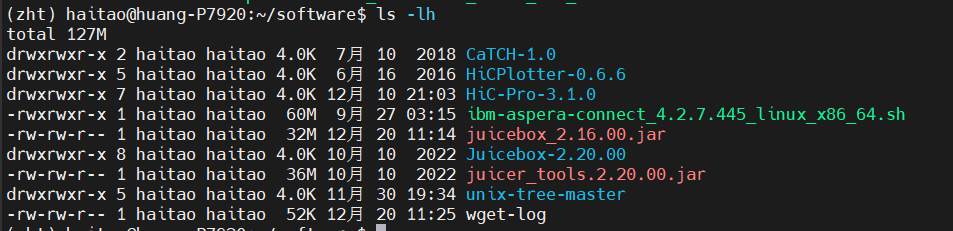
还记得我们在上一篇博客中的操作吗,使用juicer tool的工具将hic-pro转换为hic数据格式,同样的仓库里,
juicebox的jar文件在juicebox仓库中下载,使用的是juicebox_2.16的版本的jar文件(因为juicer的jar文件,无论是v2版本还是v1版本,都无法打开可视化浏览器,所以使用juicebox的jar文件),
目前尝试是:
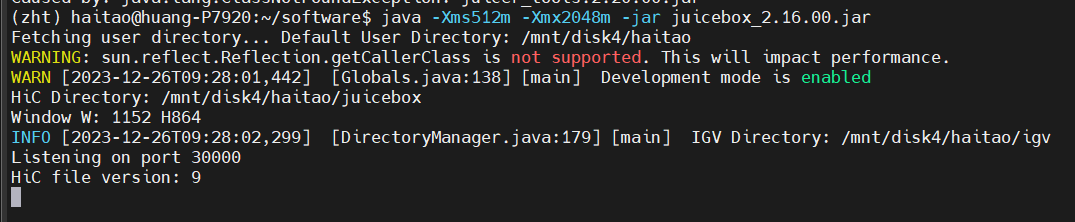
基本上在命令行中运行jar文件,都是
java -jar 一些必要的运行配置参数 jar文件 参数
参考https://github.com/aidenlab/Juicebox/issues/819,
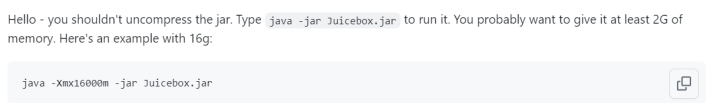

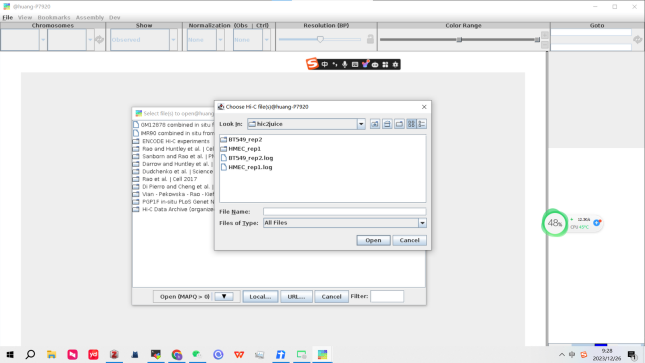
是可以打开直接linux本地上的文件的
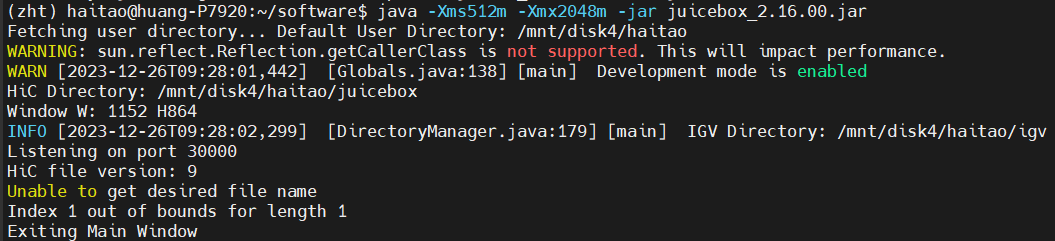 可以在对应的java -jar文件上设置最小堆2G,最大堆5G等
可以在对应的java -jar文件上设置最小堆2G,最大堆5G等
参考https://aidenlab.gitbook.io/juicebox/comparing-maps,以及生信修炼手册的https://blog.csdn.net/weixin_43569478/article/details/108079505(后面发现这篇文章锁了,可以到公众号上看看),有很多博客可以参考,看看juicebox怎么使用。
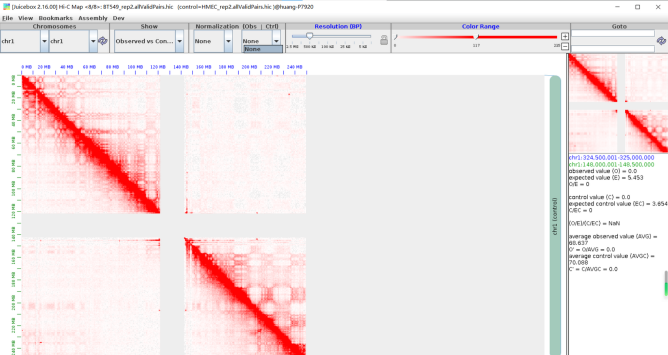
可以保存svg、pdf格式的图,比对效果是全局或者perchr上的contact的比对,
即导入case-control两个hic热图,基本上实现Obs | Ctrl效果,
注意juicebox中show list中的mode如下
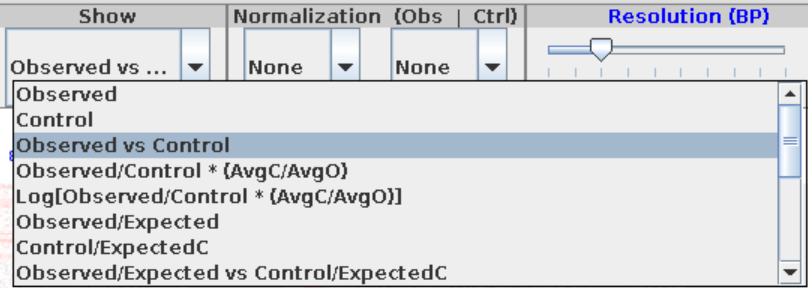
比对时:对角线之下的是实验组,对角线之上的是对照组,
实验之后能够在对角线上展示比对效果的只有下面这2种模式:
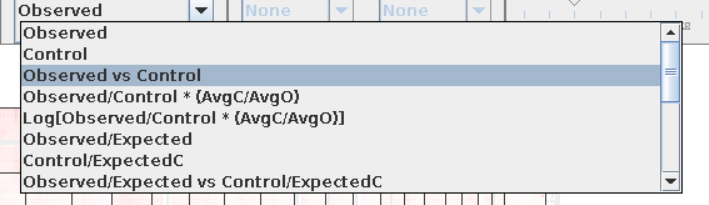
前者能够在全局+perchr上显示比对效果
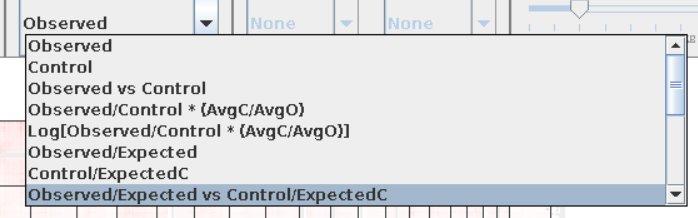
后者只能在perchr上显示效果,
除了前3列,其他list中的mode需要注意其意义。
在Chromosomes一栏中,两列都选择17号染色体,代表交互矩阵的行和列都是17号染色体,然后点击右边的刷新按钮
通过Normalization工具调整归一化方法;通过Resolution工具调整分辨率;通过Goto工具调整显示的染色体区域。
导入注释文件,通过View->Show Annotation Panel可以打开注释文件面板,注释信息分为1D和2D的两种,1D的主要指的是chip_seq,RNA_seq等信息,2D指的是TAD,染色质环等信息,通过Load ENCODE可以导入ENCODE数据库中的一维注释
每个导入的track的颜色等配置可以在面板中进行调整
通过Load Loops/Domain可以导入2D的注释信息
黄色区域标记的是TAD, 浅蓝色区域标记的是染色质环,调整好之后的图片也可以导出成PDF或者SVG格式。
除了juicebox之外,其他可以用于hic数据可视化的工具有:higlass,hicplotter等,
对应软件安装先去官方github,或者bioconda看看,或者直接在conda中search并且install;
3,相关性分析(相关性热图): 用于检测每个样本中重复rep之间的相似性
参考<https://github.com/MaybeBio/UO_project_2021/tree/main/02_visualize_replicates>
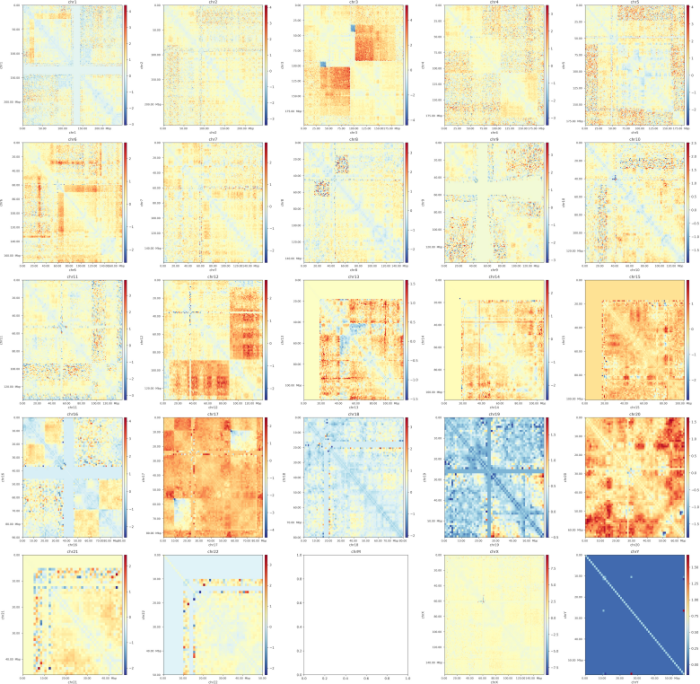
(1)计算校正分层系数SCC:
参考上面脚本StratumAdjustedCorrelationCoefficients或MDS,或参考PDXHiC_supplemental/Fig4_HiC_misc的

本人操作如下:
安装hicrep(https://github.com/dejunlin/hicrep)),对输入mcool文件,利用脚本HiCrep.sh(https://github.com/MaybeBio/HiC_pipeline/blob/main/pipeline/hic%20%E5%9F%BA%E7%A1%80pipeline%E9%AA%A8%E6%9E%B6/HiCrep.sh,参考PDXHiC_supplemental/Fig4_HiC_misc
/01_HiCrep.sh修改)获取相似性矩阵文件;
使用脚本hicrep_heatmap.R(https://github.com/MaybeBio/HiC_pipeline/blob/main/pipeline/hic%20%E5%9F%BA%E7%A1%80pipeline%E9%AA%A8%E6%9E%B6/hicrep_heatmap.R,参考https://github.com/MaybeBio/UO_project_2021/tree/main/02_visualize_replicates修改)处理相似性矩阵文件,用于绘制热图;
效果大概如下:

(2)计算MDS:
(3)计算PCA:
参考https://github.com/MaybeBio/UO_project_2021/tree/main/02_visualize_replicates/PCA
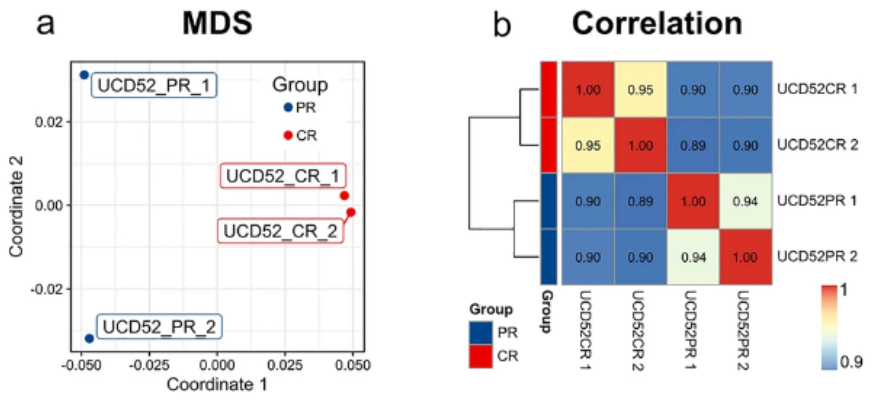
(4)另外整体上绘制相似性/可重复性图可参考https://github.com/MaybeBio/PDXHiC_supplemental/tree/main/Fig4_HiC_misc中的02_replicates_HiCrep.Rmd+03_replicates_correlation.Rmd(看md描述),效果如下:
4,绘制距离衰减曲线(Distance Decay曲线,类似于RCP,基因组互作-距离曲线):
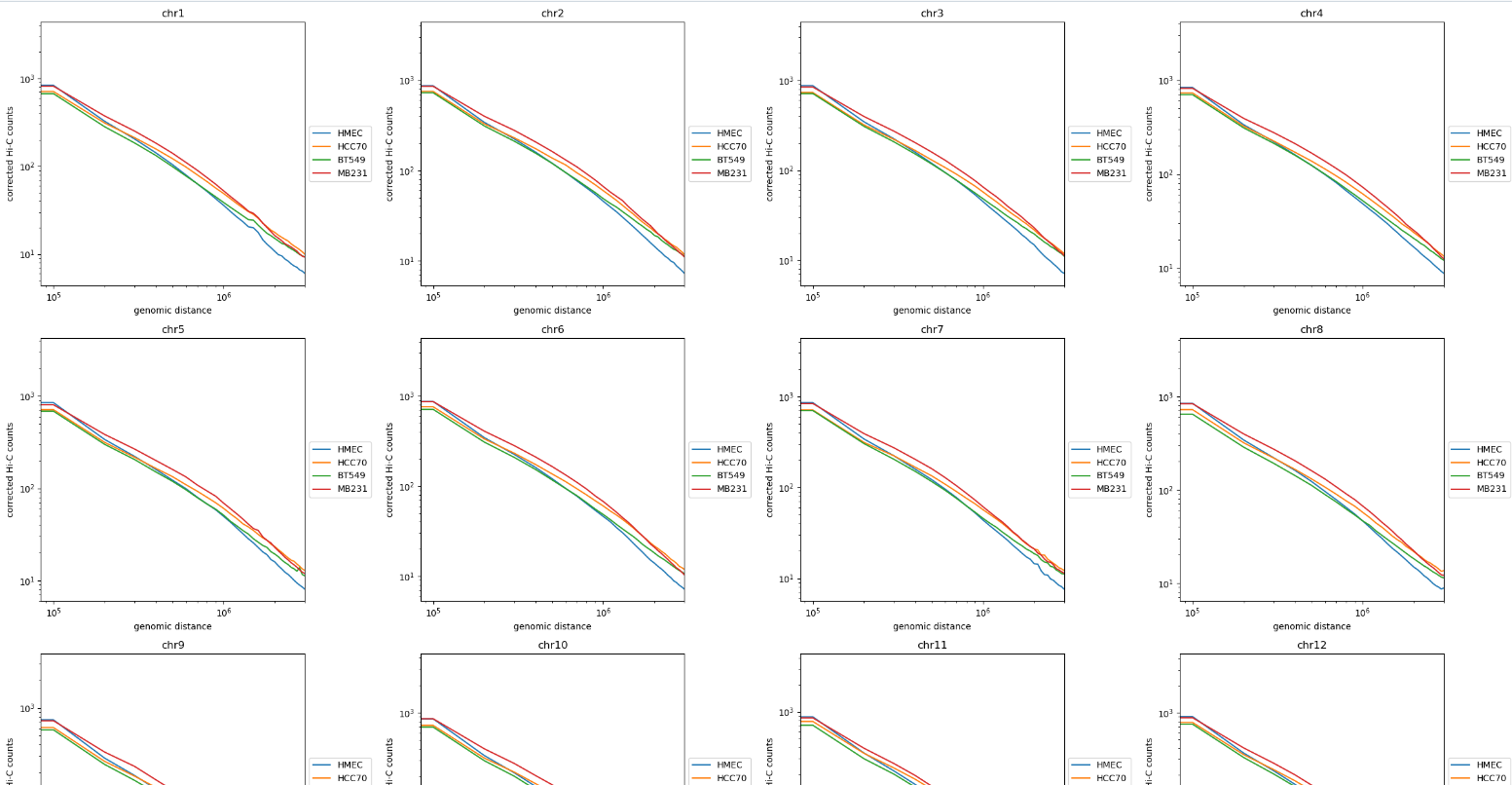
(1)使用hicexplorer中的hicPlotDistVsCounts函数:
参考https://github.com/MaybeBio/PDX-HiC_processingScripts/blob/main/7.hicPlotDistVsCounts.sh,
以及https://github.com/MaybeBio/UO_project_2021/tree/main/02_visualize_replicates/distance_decay,
效果如下:
(2)使用GENOVA中的RCP(The Relative Contact Probability)函数:
参考https://github.com/MaybeBio/PDXHiC_supplemental/blob/main/Fig4_HiC_misc/04_distance.Rmd
效果如下:
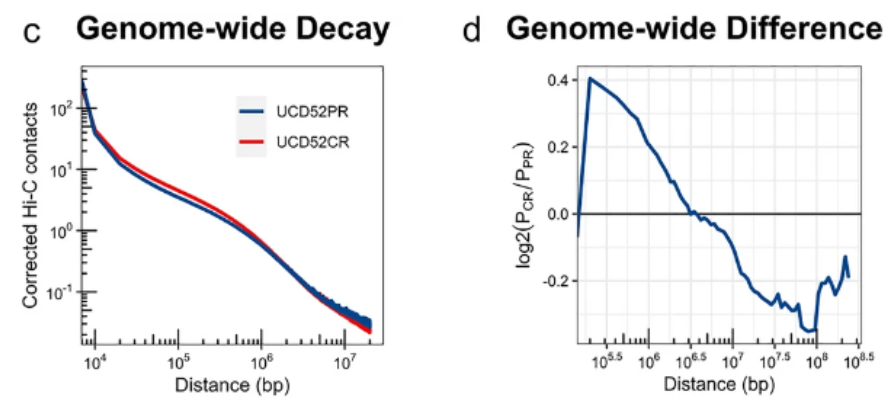
如果不使用脚本修饰,直接使用RCP函数的话,本人效果如下:
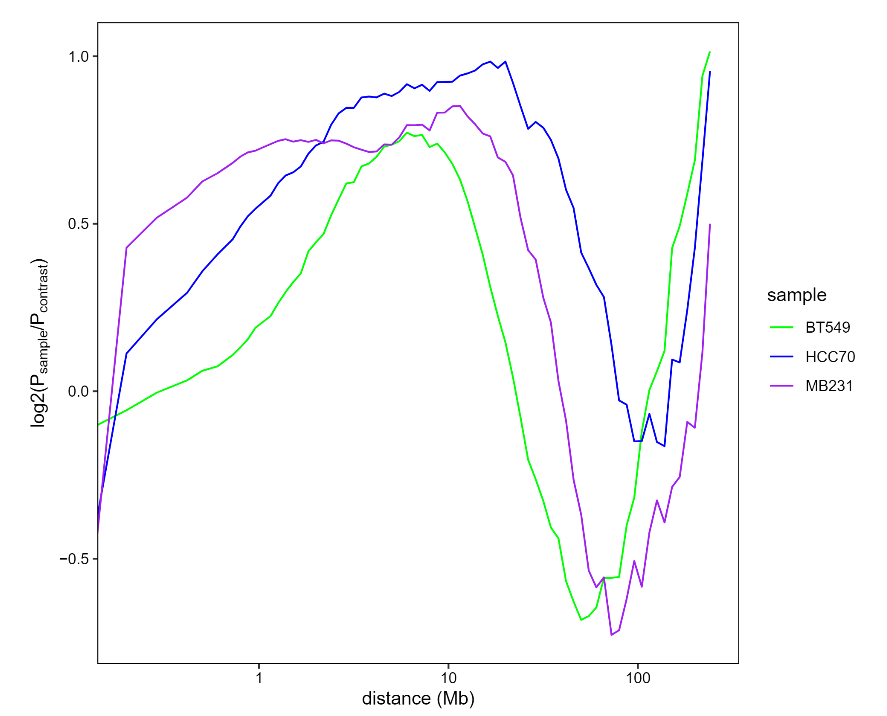
(3)其他hic下游处理软件、R包1都能够绘制RCP类似曲线:比如HITC包(也是hic-pro官方的下游分析包)
5,其他hic全局3D基因组feature,即其他hic上游数据分析模块:
(1)参考GENOVA或hicexplorer中有什么可以用于上游分析的模块
(2)参考其他CNS文献分析hic数据的,可以扩展延伸出3.1、3.2文章等















 770
770

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









