







意义
去除批次效应(batch effect removal)在组学数据分析中是一个非常重要的步骤,其主要原因可以归纳为以下几点:
一、提高数据质量
-
减少技术变异:批次效应主要是由于实验条件、操作人员、试剂批次、仪器状态等非生物因素导致的系统性偏差。这些技术变异会掩盖或模糊真实的生物学变异,影响数据的准确性和可靠性。
-
提升数据一致性:不同批次之间的数据可能由于批次效应而产生系统性偏差,导致数据在不同批次间不可比。去除批次效应可以消除这种偏差,提高数据的一致性和可比性。
二、确保分析结果的准确性和可靠性
-
改进统计分析:批次效应可能会掩盖或扭曲真实的生物学变异,使得统计分析的结果不准确。去除批次效应后,统计分析的可靠性会得到提高,能够更准确地反映生物学上的差异。
-
增强生物学解释:去除批次效应后,可以更准确地识别和解释生物学上有意义的变异,如基因表达的差异、蛋白质丰度的变化等。这对于深入理解生物学过程和疾病机制具有重要意义。
三、提高研究复现性
去除批次效应有助于提高研究的复现性。因为去除了实验条件变化带来的影响,使得其他研究者可以在相似的条件下复现实验结果,从而验证和扩展研究成果。
四、促进数据整合
-
跨实验室或跨平台数据整合:在进行跨实验室或跨平台的数据整合时,去除批次效应可以提高数据的兼容性,使得来自不同来源的数据可以进行有效的比较和整合。
-
扩大样本量:通过去除批次效应,可以将多个批次的数据合并为一个数据集,从而扩大样本量,提高统计功效和发现生物学差异的能力。
五、具体方法
去除批次效应的方法多种多样,包括但不限于ComBat(基于经典贝叶斯的分析方法)、limma、SVA等。这些方法可以对每个样本的基因表达进行调整,以消除由于批次效应引起的差异。此外,针对单细胞RNA测序数据,还有Harmony、SeuratV3等专门的方法用于去批次处理。
下面对多个数据集的基因表达芯片数据使用ComBat包进行举例:
数据准备
基因表达矩阵
exp <- cbind(GSE66099,GSE65682,GSE69528,GSE13015,GSE69686)
> dim(exp)
#[1] 7215 1404
> exp[1:4,1:4]
# GSM1617492 GSM1617493 GSM1617494 GSM1617495
#AAK1 3.771851 3.687045 3.618850 3.679106
#AAMP 4.717504 4.610980 5.362452 4.984403
#AARS 5.703946 5.529675 5.514384 5.707664
#AARS2 3.498378 3.391538 3.959782 4.011685临床信息
(在临床信息里的详情实在太多了,一般有用的是Normal和Tumor分组)
info <- rbind(g_66099,g_65682,g_69528,g_13015,g_69686) # 1393 3
rownames(info) <- info$X # 把样本名称作为行索引
info <- info[,-1] # 删除第一列
table(info$type) ## 查看数据分组,Normal:154,Sepsis:1250
#Normal Sepsis
# 154 1250
> table(info$ID) ## 查看各数据集数量
#GSE13015 GSE65682 GSE66099 GSE69528 GSE69686
# 39 802 276 138 149
> info[1:4,]
# type ID
#GSM1617492 Normal GSE66099
#GSM1617493 Normal GSE66099
#GSM1617494 Normal GSE66099
#GSM1617495 Normal GSE66099保证样本顺序一致
# 表达数据和临床数据顺序一致,方便增删
identical(rownames(info), colnames(exp))
#[1] TRUE数据初探索
在处理之前一般都会看一下数据的分布情况,可以绘制PCA或箱型图
按照分组信息分组
library(tinyarray)
# 按照type分组信息进行绘制
draw_pca(exp = exp, group_list = factor(info$type))

图中的交集部分太多,说明数据在不同分组下差别不大,好的数据在不同类型下的区别是很明显的,能正常分开。
按照批次信息分组
# 按照不同批次的数据集进行绘制
draw_pca(exp = exp, group_list = factor(info$ID)) 
在图中可以看出不同批次的数据表达水平差异很明显,说明是有必要去除批次效应,消除批次带来的影响。
去除批次效应
library(sva)
expr_combat <- ComBat(dat = exp, batch = info$ID)
## Found5batches
## Adjusting for0covariate(s) or covariate level(s)
## Standardizing Data across genes
## Fitting L/S model and finding priors
## Finding parametric adjustments
## Adjusting the Data
# 查看去除批次后的数据
expr_combat[1:4,1:4]
# GSM1617492 GSM1617493 GSM1617494 GSM1617495
# AAK1 4.881798 4.751416 4.646573 4.739211
# AAMP 4.535422 4.452894 5.035089 4.742200
# AARS 4.539075 4.409302 4.397915 4.541844
# AARS2 3.189297 3.074617 3.684559 3.740271
expr_combat <- as.data.frame(expr_combat)
# 再画图看看
draw_pca(exp = expr_combat, group_list = factor(info$type))
效果很明显,与没去批次之前差别很大,但是还有小部分Normal和Sepsis的混在一起。
在ComBat里面有个mod选项,可以用来指定感兴趣的分组(这里就是normal和Sepsis),告诉函数不要把本来的分组信息给整没了。
mod <- model.matrix(~factor(info$group))
expr_combat <- ComBat(dat = exp, batch = info$ID,mod = mod)
# Found5batches
# Adjusting for1covariate(s) or covariate level(s)
# Standardizing Data across genes
# Fitting L/S model and finding priors
# Finding parametric adjustments
# Adjusting the Data
draw_pca(exp = expr_combat, group_list = factor(info$type))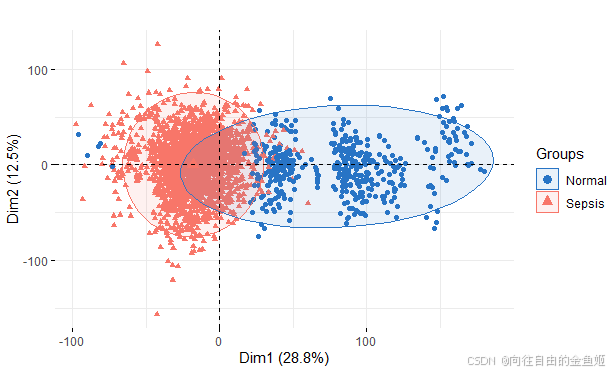
跟上一张图相比,Normal与Sepsis区别更明显了。
综上所述,去除批次效应是组学数据分析中不可或缺的一步,它对于提高数据质量、确保分析结果的准确性和可靠性、提高研究复现性以及促进数据整合都具有重要意义。

















 486
486

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









