







为什么需要去除批次效应?
批次效应是指样本在不同批次处理及测量的情况下引入与生物学情况不相关的技术误差,比如不同试剂耗材,不同操作者,不同的实验时间等。
正是因为这些非生物学的因素存在就有可能会导致我们的结果偏离真实的情况,那么实际分析的过程中研究者应当评估是否存在批次效应,并决定是否要进行去批次处理。
值得注意的是,即使使用了所谓的去批次效应的工具,批次效应仍不能被完全消除,只是尽可能的减少了批次带来的干扰!
载入数据并可视化探索
1.加载R包
rm(list = ls())
library(bladderbatch)
library(sva)
library(ggplot2)
library(FactoMineR) # PCA函数
library(factoextra) # fviz_pca_ind函数
library(pheatmap) # pheatmap函数
library(sva) # 用于combat和combat_seq
library(limma) # 用于removeBatchEffect
proj = "bladderbatch"
2.加载示例数据
data(bladderdata)
pheno = pData(bladderEset)
head(pheno)[1:4,1:4]
# sample outcome batch cancer
# GSM71019.CEL 1 Normal 3 Normal
# GSM71020.CEL 2 Normal 2 Normal
# GSM71021.CEL 3 Normal 2 Normal
# GSM71022.CEL 4 Normal 3 Normal
exprSet = exprs(bladderEset)
head(exprSet)[1:4,1:4]
# GSM71019.CEL GSM71020.CEL GSM71021.CEL GSM71022.CEL
# 1007_s_at 10.115170 8.628044 8.779235 9.248569
# 1053_at 5.345168 5.063598 5.113116 5.179410
# 117_at 6.348024 6.663625 6.465892 6.116422
# 121_at 8.901739 9.439977 9.540738 9.254368
batch = pheno$batch
head(batch)
# [1] 3 2 2 3 3 3
3.原始数据可视化
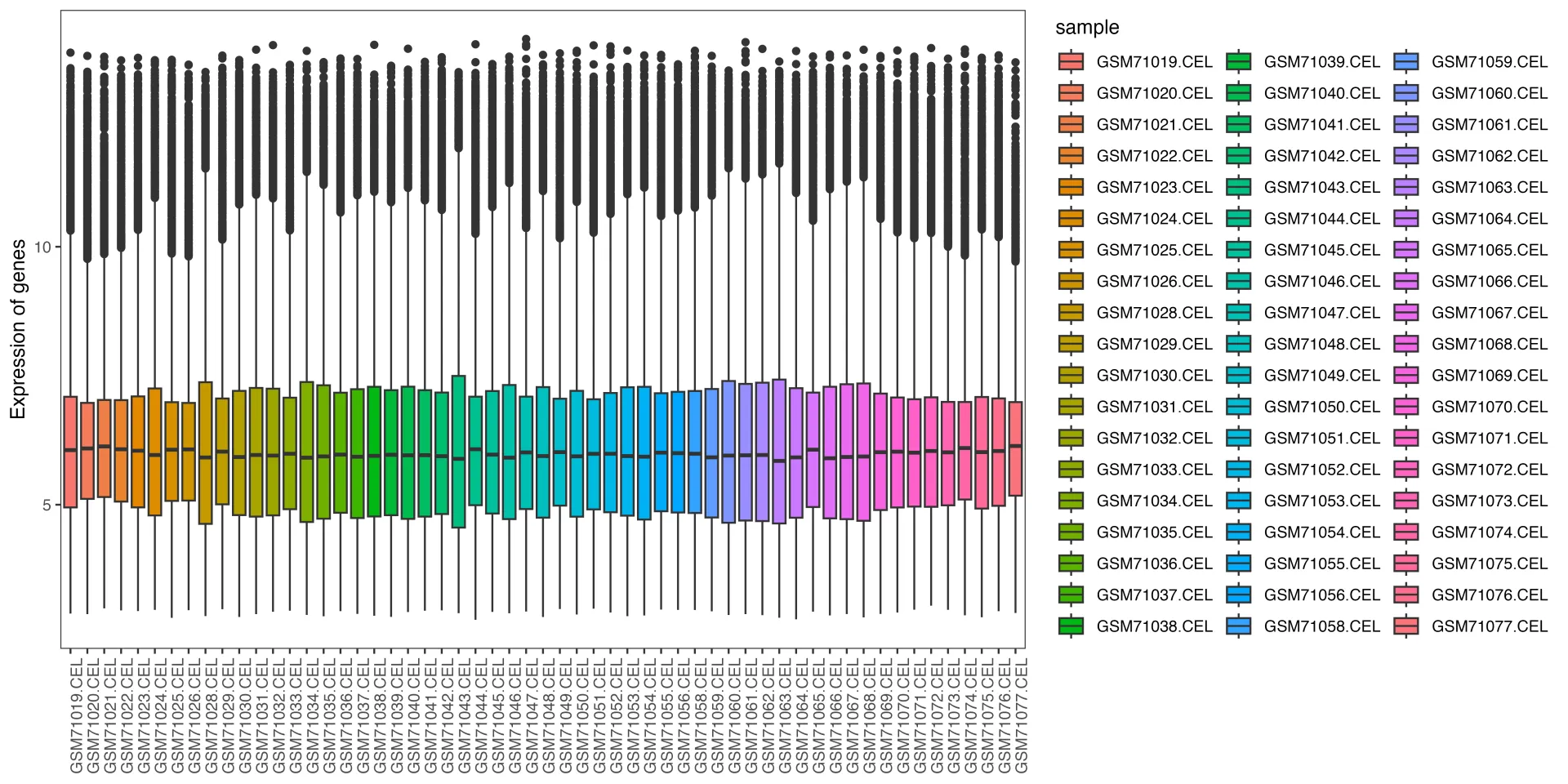
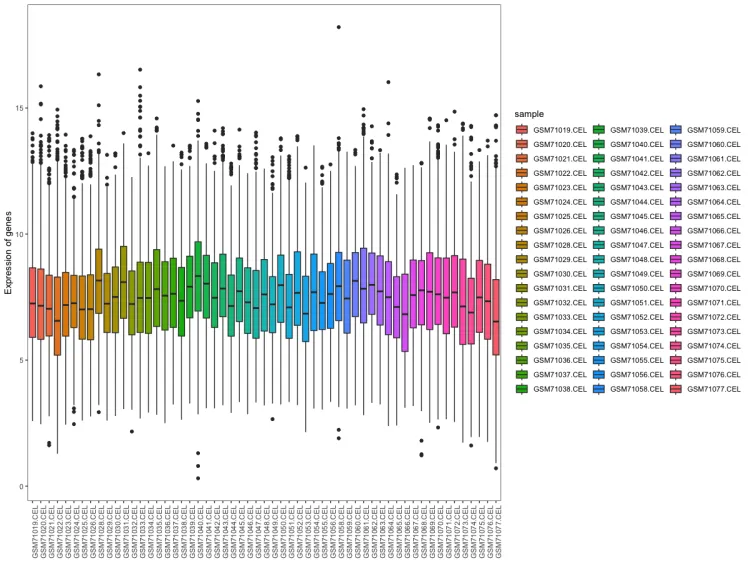
# 样本表达总体分布-箱式图--------------------------------------
mythe <- theme_bw() + theme(panel.grid.major=element_blank(),
panel.grid.minor=element_blank())
# 构造绘图数据
data <- data.frame(expression=c(exprSet),
sample=rep(colnames(exprSet),
each=nrow(exprSet)))
head(data)
dim(data)
# 箱线图
p <- ggplot(data = data, aes(x=sample, y=expression, fill=sample))
p1 <- p + geom_boxplot() +
mythe +
theme(axis.text.x = element_text(angle = 90)) +
xlab(NULL) + ylab("Expression of genes") #+ scale_fill_nejm()
p1
ggsave("1.sample_boxplot.png", plot = p1, dpi = 600, width = 12, height = 6, units = "in")
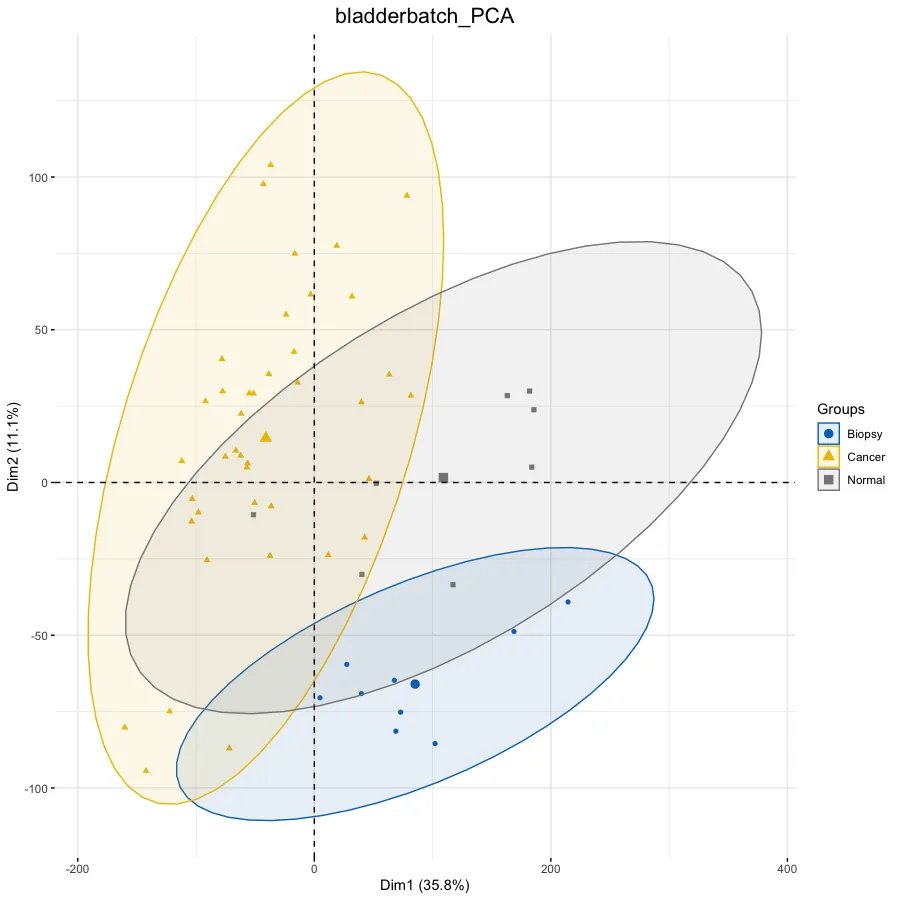
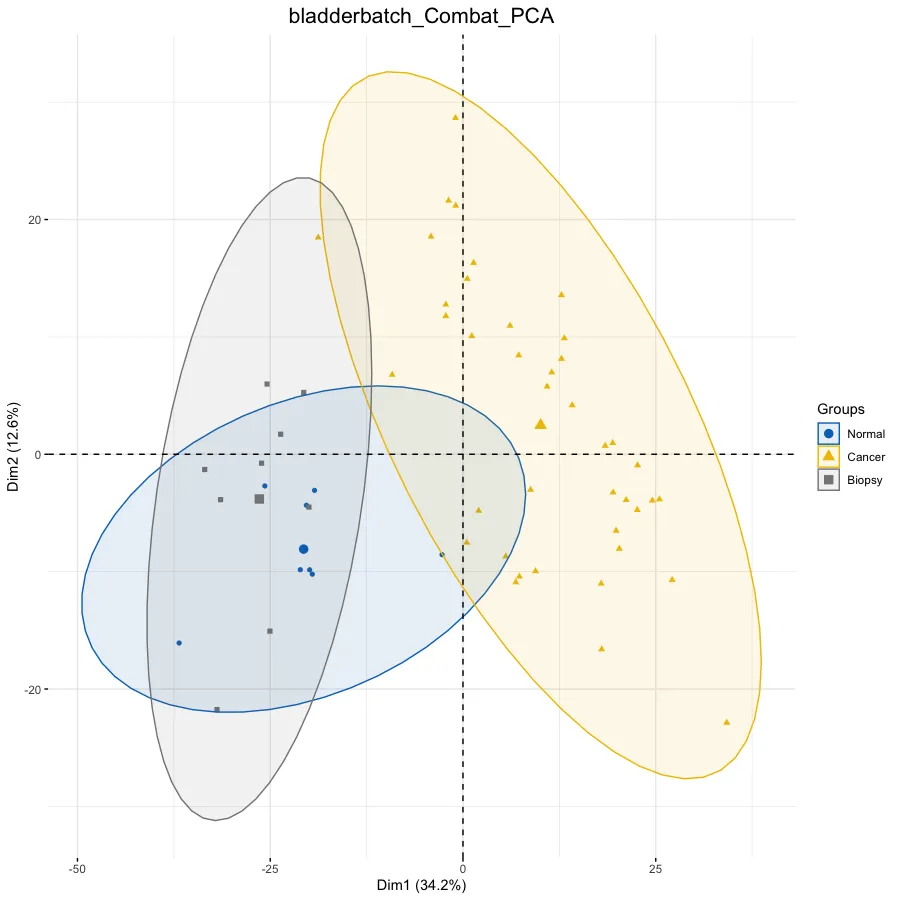
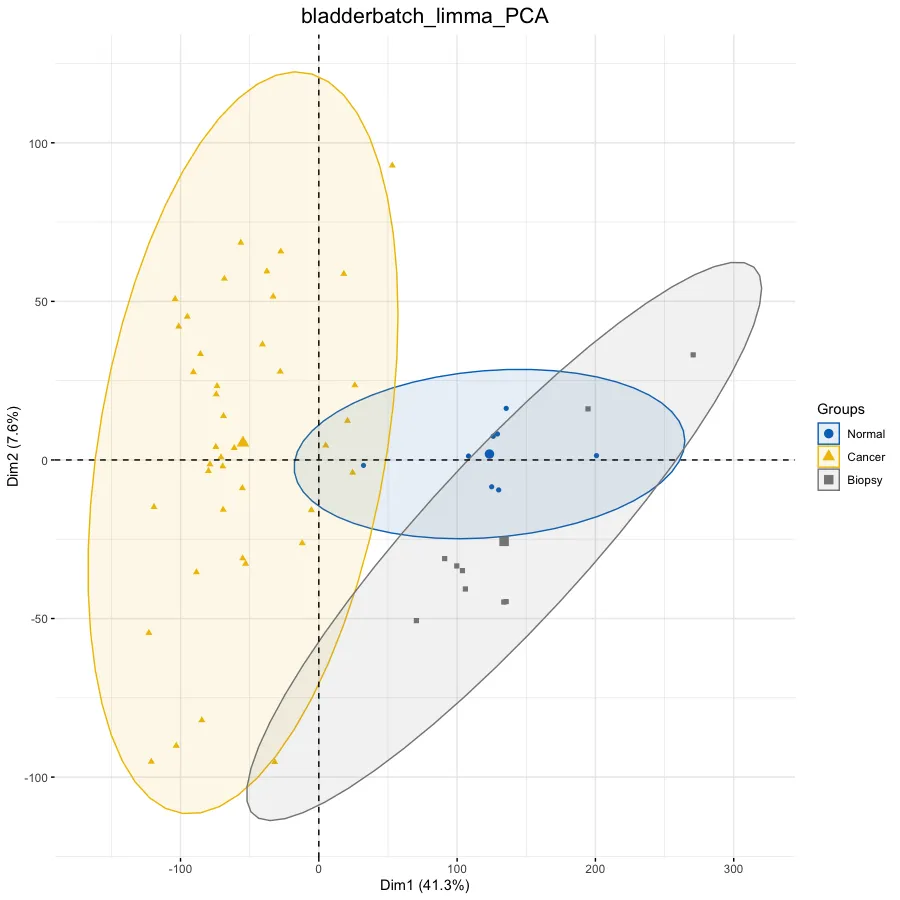
# PCA分析--------------------------------------------------
# PCA图时要求是行名时样本名,列名时探针名,因此此时需要转换
exp = t(exprSet)
# 将matrix转换为data.frame
exp = as.data.frame(exp)
dim(exp)
exp[1:4, 1:4]
# 1007_s_at 1053_at 117_at 121_at
# GSM71019.CEL 10.115170 5.345168 6.348024 8.901739
# GSM71020.CEL 8.628044 5.063598 6.663625 9.439977
# GSM71021.CEL 8.779235 5.113116 6.465892 9.540738
# GSM71022.CEL 9.248569 5.179410 6.116422 9.254368
# 添加分组信息
ac <- data.frame(
row.names = rownames(exp),
Group = pheno$cancer)
# 设置图片标题
pro = proj
this_title <- paste0(pro, '_PCA')
# 绘图
dat.pca <- PCA(exp, graph = FALSE)
p.pca <- fviz_pca_ind(dat.pca,
# 只显示点而不显示文本,默认都显示
geom.ind = "point", #c( "point", "text" ) / "point",
# geom.ind = 'text',
# 设定分类种类
col.ind = ac$Group, # color by groups
# 设定颜色
palette = "jco", # jco/Dark2
# 添加椭圆
addEllipses = TRUE, # Concentration ellipses
# 添加图例标题
legend.title = "Groups") +
ggtitle(this_title) +
theme(plot.title = element_text(size=16, hjust = 0.5))
p.pca
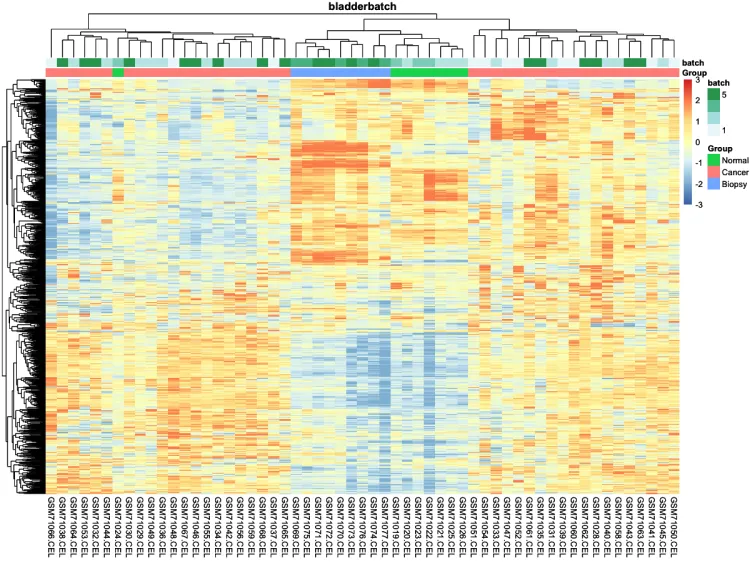
# 热图——————————————————————————————————
# 设置图片标题
pro = proj
this_title <- paste0(pro, '_PCA')
# 取方差top1000的基因绘制热图
# 因为基因表达矩阵过大,选择性进行基因过滤
# apply按行('1'是按行取,'2'是按列取)取每一行的方差,从小到大排序,取最大的1000个
cg = names(tail(sort(apply(exprSet, 1, sd)), 1000))
head(cg)
exprSet = exprSet[cg, ]
exprSet[1:4, 1:4]
dim(exprSet)
# [1] 1000 57
# 绘制热图-check
pheatmap(exprSet, show_colnames = F, show_rownames = F)
# 数据标准化化及绘图
# scale()函数将每一列看作一个样本数据
n = t(scale(t(exprSet)))
n[n>2]=2
n[n< -2]= -2
head(n)
# 标准化后的数据绘制热图-check
pheatmap(n, show_colnames = F, show_rownames = F)
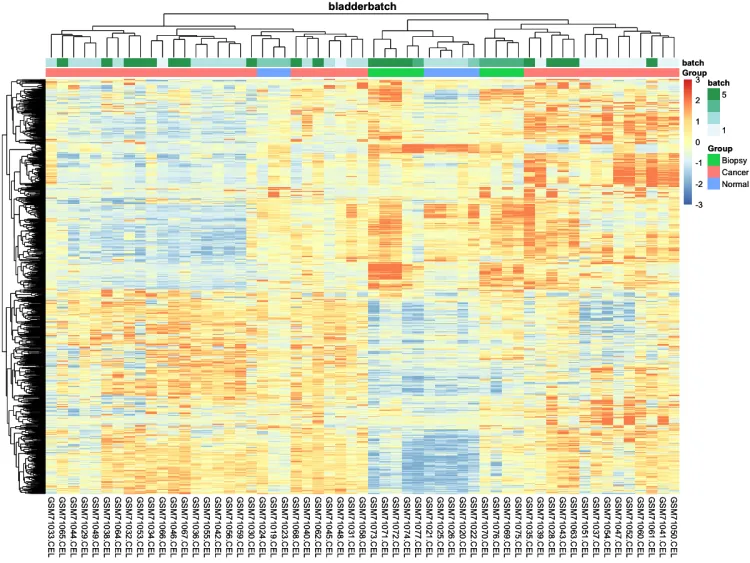
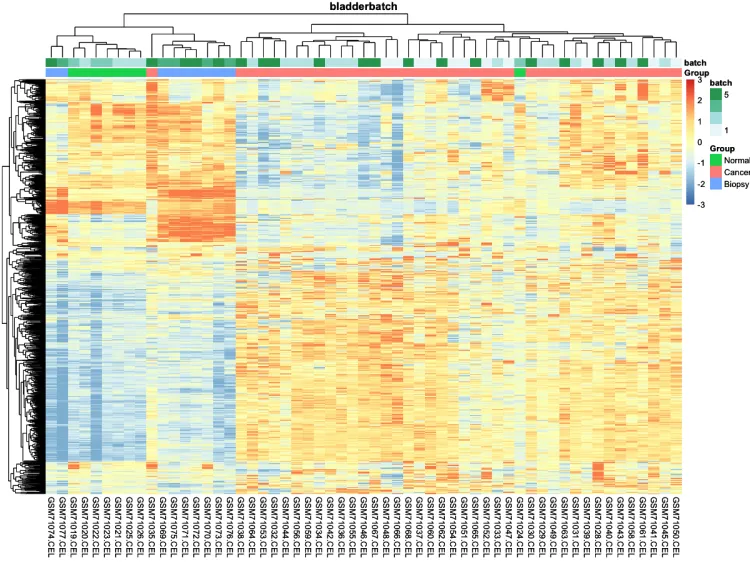
# 添加分组信息
ac <- data.frame(
row.names = colnames(n),
Group = pheno$cancer,
batch = pheno$batch)
# 绘制添加分组信息热图
p.heatmap <- pheatmap::pheatmap(n,
main = pro, # 设置图片标题
annotation_col = ac,# 添加列分组信息
show_colnames = T,# 不展示列名
show_rownames = F, # 不展示行名
breaks = seq(-3, 3, length.out = 100)) # 设置间隔
p.heatmap
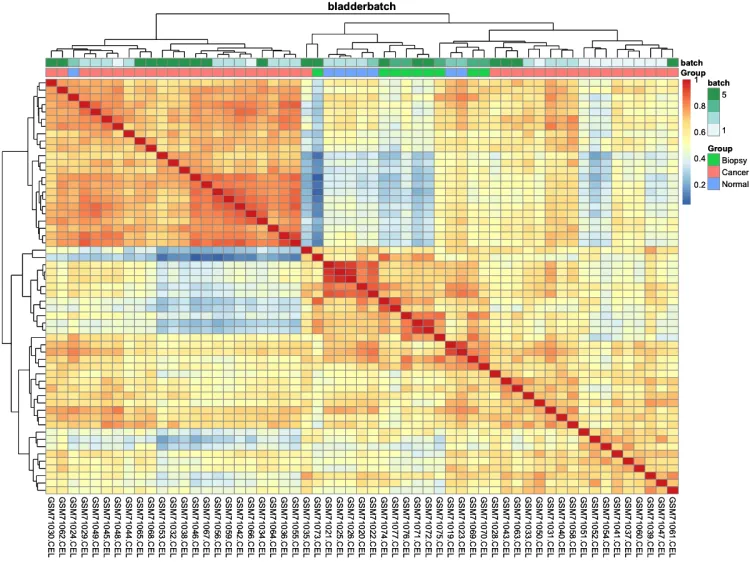
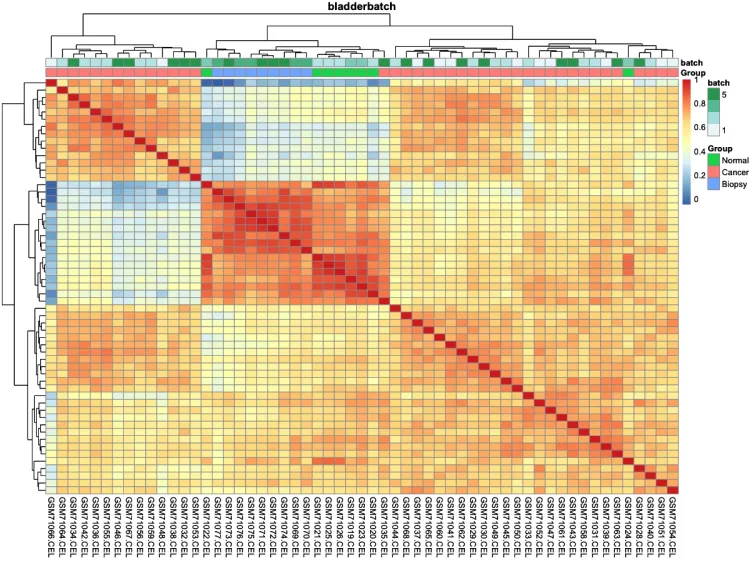
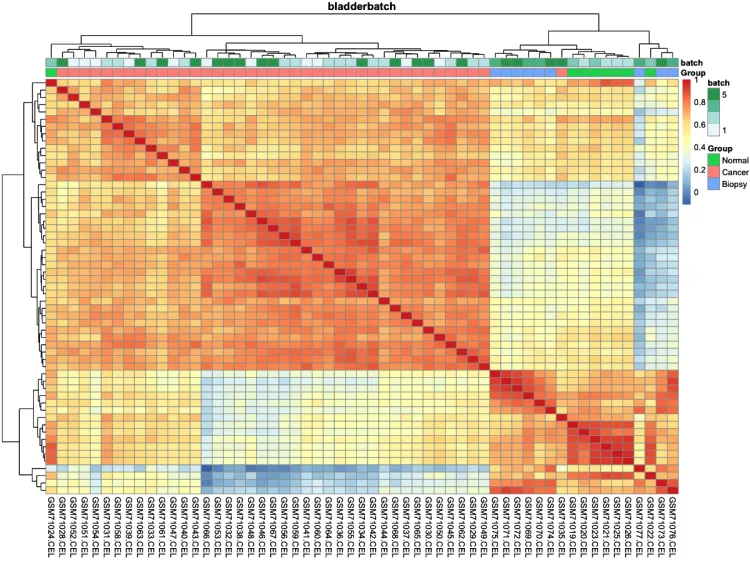
# 绘制组间相关性热图(样本-样本)-check
pheatmap::pheatmap(cor(exprSet, method = 'spearman'))
# 添加分组信息
ac = data.frame(row.names = colnames(exprSet),
Group = pheno$cancer,
batch = pheno$batch)
# 绘制组间相关性热图
p.heatmap.cluster <- pheatmap::pheatmap(cor(exprSet),
annotation_col = ac, # 添加列分组信息
display_numbers = F, # 不显示相关系数
show_rownames = F, # 不展示行名
show_colnames = T, # 不展示行名
main = pro) # 设置图片标题
p.heatmap.cluster
方法一:combat-sva

combat函数是基于贝叶斯框架下的调整数据批次效应的函数,主要适用于已经被过滤和标准化后的数据(芯片数据/非count数据)。
combat去批次-可视化
# 官方展示了三种函数设置方式
# combat_edata1 = ComBat(dat=edata, batch=batch, mod=NULL, par.prior=TRUE, prior.plots=FALSE)
# 参数方法 (par.prior=TRUE): 这种方法假设数据的批次效应可以通过正态分布的参数(均值和方差)来调整。mod=NULL: 这表示在调整批次效应时没有考虑其他协变量。这是一个纯粹的批次效应调整,不考虑如癌症状态等其他因素。
# combat_edata2 = ComBat(dat=edata, batch=batch, mod=NULL, par.prior=FALSE, mean.only=TRUE)
# 非参数方法 (par.prior=FALSE): 这种方法不假设批次效应的参数分布,而是直接估计和调整每个批次的效应,更加灵活地适应各种数据分布。只调整均值 (mean.only=TRUE):这种调整只考虑批次间的均值差异,不调整方差。适用于批次间方差相似,但均值有偏差的情况。
# combat_edata3 = ComBat(dat=edata, batch=batch, mod=mod, par.prior=TRUE, ref.batch=3)
# 包含协变量 (mod=mod): 这里的模型矩阵 mod 包括了癌症状态(cancer),这意味着调整批次效应的同时也会考虑癌症状态的影响,以防止这些生物变量的影响被误当作批次效应调整掉。指定参考批次 (ref.batch=3): 在进行批次效应调整时,将第三个批次作为参照(基准),调整其他批次的数据以匹配这个参照批次的分布。这适用于当某个批次被认为是质量最高或最标准的情况。
# 设置批次信息
batch <- pheno$batch # 批次
# 设置生物学分类,告诉函数不要把生物学差异整没了
pheno$cancer <- factor(pheno$cancer, levels = c("Normal", "Cancer", "Biopsy"))
mod <- model.matrix(~as.factor(cancer), data=pheno)
expr_combat <- ComBat(dat = exprSet, batch = batch, mod = mod,par.prior=TRUE, ref.batch=1)
可视化代码类似就不展示了
方法二:combat_seq-sva

combat-seq函数是基于二项回归分布下的调整数据批次效应的函数,主要适用于高通量测序数据获得的count数据。
combat-seq去批次
# 官方展示了三种函数设置方式
# adjusted <- ComBat_seq(count_matrix, batch=batch, group=NULL)
# group=NULL 表示没有指定生物协变量进行保留。group就是combat中的mod
# adjusted_counts <- ComBat_seq(count_matrix, batch=batch, group=group)
# group 设置了生物学分组
# cov1 <- rep(c(0,1), 4)
# cov2 <- c(0,0,1,1,0,0,1,1)
# covar_mat <- cbind(cov1, cov2)
# adjusted_counts <- ComBat_seq(count_matrix, batch=batch, group=NULL, covar_mod=covar_mat)
# cov1 和 cov2: 两个向量,表示两个不同的生物协变量。这意味着你可以在你的样本中保留两种生物学差异。
# 设置批次信息
batch <- pheno$batch
# 设置生物学分类,告诉函数不要把生物学差异整没了
mod <- pheno$cancer
# 请注意这里的exprSet数据不是count数据!!!!只是用于演示!!!!
expr_combat_seq <- ComBat_seq(exprSet, batch = batch, group = mod)
因为数据不合适,所以没有进行可视化。
方法三:removeBatchEffect-limma

removeBatchEffect函数是基于线性模型下的调整数据批次效应的函数,主要适用于芯片数据/非count的高通量数据。
removeBatchEffect去批次
# 设置批次信息
batch <- pheno$batch # 批次
# 设置生物学分类,告诉函数不要把生物学差异整没了
pheno$cancer <- factor(pheno$cancer, levels = c("Normal", "Cancer", "Biopsy"))
mod <- model.matrix(~as.factor(cancer), data=pheno)
expr_limma <- removeBatchEffect(exprSet,batch=batch,batch2=NULL,
covariates=NULL,design= mod)
# batch/batch2代表的是批次信息;covariates代表那些可能对研究结果有影响的外部变量;design是分析中不会去除与实验核心目的相关的变量,可以理解为生物学分组。
可视化代码类似就不展示了
参考资料:
1、Combat: https://rdrr.io/bioc/sva/man/ComBat.html
2、ComBat-seq: https://github.com/zhangyuqing/ComBat-seq
3、removeBatchEffect:https://rdrr.io/bioc/limma/man/removeBatchEffect.html
4、生信技能树:https://mp.weixin.qq.com/s/8fhDZiGzCna8FY49l18mtQ
5、生信菜鸟团:https://mp.weixin.qq.com/s/E2HLaym_LqJrJLKHjfpX8A
致谢:感谢曾老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -

















 531
531

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









